
A Two-Part Mixed Model for Differential Expression Analysis in Single-Cell High-Throughput Gene Expression Data



Abstract
:1. Introduction
2. Materials and Methods
2.1. The Two-Part Mixed Model for Single-Cell Gene Expression Data
2.2. Model Fitting
2.3. Testing for Differential Expression
- (1)
- Testing of the binomial part
- (2)
- Testing of the Gaussian partand the two parts can also be tested jointly, which can improve the statistical power:
- (3)
- Joint testing of the binomial and Gaussian parts
- (1)
- Fit model (4) under and generate N random samples from this model.
- (2)
- Calculate the corresponding test statistics (i.e., Wald or likelihood ratio statistics) using the above-simulated samples .
- (3)
- Estimate the p-value as , where is the test statistic (Wald or likelihood ratio) calculated from the observed data (an alternative formula is . The two formulas give almost the same results providing is large, so we use the former throughout this chapter).
- (1)
- Approximate the null distribution of test statistics ( or ) by a scaled distribution with as the scale parameter and as the degrees of freedom. The parameters and can be estimated by matching the first two moments (sample mean and variance) of test statistics under with those of [20,21]. The sample mean and variance of test statistics under can be obtained by using the above parametric bootstrap method with a smaller number of random samples.
- (2)
- Fit a two-component normal mixture distribution on , where is the p-value obtained from the above-scaled distribution for the random sample and is the standard normal CDF. The final p-values are calculated aswhere is the p-value obtained from Step (1) and is the fitted normal mixture distribution . The Satterthwaite method can estimate p-values using a smaller number of random samples than the parametric bootstrap method [20,21]. However, in our simulations, it also shows an inflated type I error rate when the sample size is small (see simulations in the next section).
3. Results
3.1. Simulation Studies
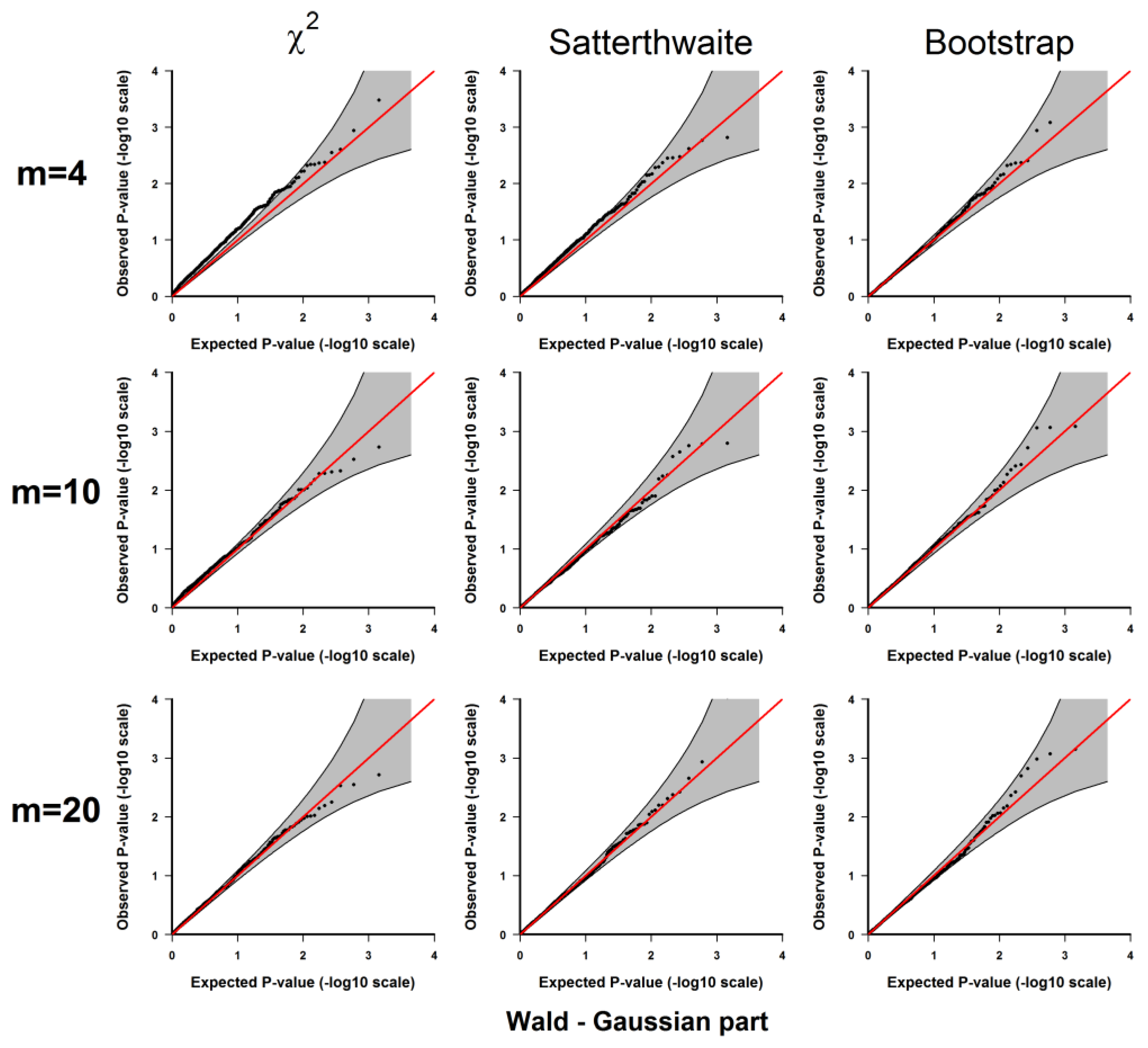
3.1.1. Evaluation of Type I Error Rates
3.1.2. Evaluation of Statistical Power
4. Application to Real-World Single-Cell Gene Expression Data
4.1. Application to an scRT-qPCR Dataset
4.2. Application to scRNA-seq Datasets
5. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | MAST | TMM | ||||
|---|---|---|---|---|---|---|
| Gaussian | Binomial | Combine | Gaussian | Binomial | Combine | |
| CD40LG | 2.33 × 10−46 | 9.87 × 10−18 | 2.82 × 10−61 | 3.73 × 10−48 | 3.53 × 10−17 | 1.72 × 10−62 |
| GAPDH | 6.60 × 10−27 | 8.44 × 10−10 | 4.04 × 10−34 | 1.78 × 10−27 | 2.30 × 10−10 | 2.82 × 10−35 |
| TNF | 1.60 × 10−03 | 7.70 × 10−22 | 1.13 × 10−22 | 3.48 × 10−03 | 1.89 × 10−22 | 7.94 × 10−23 |
| TGFB1 | 6.08 × 10−18 | 2.73 × 10−04 | 9.61 × 10−20 | 1.75 × 10−16 | 4.31 × 10−04 | 5.01 × 10−18 |
| IL2 | 1.46 × 10−03 | 4.53 × 10−18 | 3.06 × 10−19 | 2.39 × 10−03 | 3.90 × 10−18 | 2.05 × 10−19 |
| IL16 | 2.21 × 10−01 | 9.21×10−18 | 2.93 × 10−16 | 4.90 × 10−02 | 4.74 × 10−18 | 2.42 × 10−17 |
| IL2Rg | 6.80×10−08 | 1.97 × 10−10 | 3.72 × 10−16 | 3.11 × 10−09 | 2.08 × 10−11 | 2.06 × 10−18 |
| CXCR4 | 7.89 × 10−04 | 3.94 × 10−14 | 1.33 × 10−15 | 5.20 × 10−04 | 1.71 × 10−14 | 3.51 × 10−16 |
| CCR7 | 3.60 × 10−01 | 8.61 × 10−17 | 4.88 × 10−15 | 4.20 × 10−01 | 8.38 × 10−17 | 3.54 × 10−15 |
| CD3d | 3.69 × 10−06 | 1.67 × 10−10 | 1.65 × 10−14 | 7.22 × 10−07 | 2.33 × 10−10 | 3.61 × 10−15 |
| IL2Ra | 9.09 × 10−03 | 5.14 × 10−13 | 2.08 × 10−13 | 1.18 × 10−03 | 1.09 × 10−12 | 6.11 × 10−14 |
| CD69 | 7.99 × 10−06 | 2.06 × 10−09 | 4.00 × 10−13 | 3.34 × 10−06 | 1.89 × 10−09 | 1.80 × 10−13 |
| IL10 | 1.75 × 10−01 | 3.88 × 10−14 | 5.74 × 10−13 | 3.25 × 10−02 | 5.85 × 10−14 | 1.44 × 10−13 |
| FASLG | 6.47 × 10−02 | 1.60 × 10−12 | 3.73 × 10−12 | 3.06 × 10−02 | 1.33 × 10−12 | 3.32 × 10−12 |
| IL7R | 1.23 × 10−06 | 2.18 × 10−06 | 5.23 × 10−11 | 9.05 × 10−08 | 1.68 × 10−06 | 4.57 × 10−12 |
| IL6ST | 1.13 × 10−03 | 4.49 × 10−09 | 1.21 × 10−10 | 1.14 × 10−04 | 4.45 × 10−08 | 1.12 × 10−10 |
| SLAMF1 | 3.45 × 10−02 | 4.78 × 10−10 | 5.56 × 10−10 | 1.05 × 10−02 | 2.95 × 10−10 | 9.47 × 10−11 |
| IFNg | 2.66 × 10−05 | 1.47 × 10−06 | 7.06 × 10−10 | 2.39 × 10−05 | 1.85 × 10−06 | 5.83 × 10−10 |
| CD109 | 8.97 × 10−01 | 8.36 × 10−10 | 8.06 × 10−09 | 7.76 × 10−01 | 7.10 × 10−10 | 7.65 × 10−09 |
| TNFRSF9 | 3.20 × 10−01 | 2.38 × 10−09 | 1.44 × 10−08 | 2.13 × 10−01 | 1.45 × 10−09 | 8.35 × 10−09 |
| DPP4 | 2.96 × 10−01 | 1.35 × 10−09 | 1.91 × 10−08 | 4.66 × 10−02 | 2.03 × 10−08 | 2.38 × 10−08 |
| ICOS | 2.41 × 10−05 | 6.80 × 10−05 | 2.53 × 10−08 | 8.93 × 10−05 | 5.14 × 10−04 | 4.77 × 10−07 |
| CD28 | 2.14 × 10−01 | 3.34 × 10−07 | 1.88 × 10−06 | 3.67 × 10−01 | 1.17 × 10−06 | 9.26 × 10−06 |
| CD4 | 1.06 × 10−01 | 1.47 × 10−05 | 2.46 × 10−05 | 1.04 × 10−01 | 3.88 × 10−05 | 6.66 × 10−05 |
| CD27 | 1.94 × 10−01 | 2.86 × 10−05 | 8.73 × 10−05 | 1.01 × 10−01 | 5.68 × 10−06 | 1.20 × 10−05 |
| CD48 | 4.55 × 10−01 | 1.69 × 10−05 | 1.59 × 10−04 | 3.52 × 10−01 | 1.46 × 10−06 | 2.14 × 10−05 |
| SLAMF5 | 5.39 × 10−02 | 3.28 × 10−04 | 1.89 × 10−04 | 3.34 × 10−02 | 4.43 × 10−04 | 1.14 × 10−04 |
| CTSD | 3.11 × 10−03 | 7.58 × 10−03 | 2.14 × 10−04 | 3.59 × 10−04 | 8.01 × 10−03 | 2.68 × 10−05 |
| CD5 | 5.47 × 10−01 | 3.30 × 10−05 | 3.65 × 10−04 | 3.80 × 10−01 | 7.13 × 10−06 | 4.89 × 10−05 |
| TBX21 | 1.71 × 10−01 | 1.97 × 10−04 | 4.06 × 10−04 | 1.17 × 10−01 | 8.15 × 10−05 | 1.10 × 10−04 |
| CSF2 | 6.55 × 10−01 | 2.46 × 10−04 | 5.40 × 10−04 | 1.00 | 2.09 × 10−04 | 5.13 × 10−04 |
| CD3g | 9.95 × 10−01 | 1.17 × 10−05 | 6.03 × 10−04 | 8.13 × 10−01 | 7.87 × 10−07 | 3.11 × 10−05 |
| TIA1 | 1.70 × 10−02 | 1.29 × 10−02 | 1.64 × 10−03 | 9.89 × 10−03 | 4.89 × 10−03 | 3.84 × 10−04 |
| CD45 | 2.94 × 10−01 | 8.41 × 10−04 | 2.56 × 10−03 | 1.72 × 10−01 | 2.48 × 10−03 | 3.63 × 10−03 |
| PECAM1 | 3.68 × 10−02 | 1.21 × 10−02 | 3.14 × 10−03 | 7.98 × 10−03 | 9.12 × 10−03 | 5.55 × 10−04 |
| NT5E | 5.96 × 10−01 | 2.21 × 10−03 | 6.24 × 10−03 | 3.77 × 10−01 | 1.28 × 10−03 | 3.82 × 10−03 |
| LIF | 7.70 × 10−01 | 3.60 × 10−03 | 1.17 × 10−02 | 6.83 × 10−01 | 1.00 | 7.22 × 10−01 |
| FOXP3 | 8.77 × 10−03 | 2.03 × 10−01 | 1.21 × 10−02 | 3.65 × 10−03 | 2.02 × 10−01 | 6.33 × 10−03 |
| TIMP1 | 2.18 × 10−02 | 1.26 × 10−01 | 1.62 × 10−02 | 4.23 × 10−02 | 7.55 × 10−02 | 2.13 × 10−02 |
| CTLA4 | 3.26 × 10−01 | 8.50 × 10−03 | 1.92 × 10−02 | 4.15 × 10−02 | 1.70 × 10−02 | 5.31 × 10−03 |
| FAS | 7.88 × 10−01 | 4.45 × 10−03 | 3.49 × 10−02 | 4.21 × 10−01 | 4.76 × 10−03 | 1.21 × 10−02 |
| RORC | 8.99 × 10−01 | 1.13 × 10−02 | 3.61 × 10−02 | 9.52 × 10−01 | 7.71 × 10−03 | 2.06 × 10−02 |
| CCR2 | 3.11 × 10−02 | 2.07 × 10−01 | 3.73 × 10−02 | 1.42 × 10−04 | 5.63 × 10−01 | 9.61 × 10−04 |
| BCL2 | 2.34 × 10−01 | 3.77 × 10−02 | 4.15 × 10−02 | 1.56 × 10−01 | 1.00 | 1.51 × 10−01 |
| PRDM1 | 1.36 × 10−02 | 5.71 × 10−01 | 5.18 × 10−02 | 2.11 × 10−03 | 7.40 × 10−01 | 1.85 × 10−02 |
| CCL3 | 1.48 × 10−01 | 1.01 × 10−01 | 6.68 × 10−02 | 4.16 × 10−01 | 1.00 | 3.36 × 10−01 |
| CCL2 | 8.07 × 10−02 | 1.00 | 8.07 × 10−02 | 2.14 × 10−01 | 1.00 | 3.05 × 10−01 |
| IL8 | 1.03 × 10−01 | 2.06 × 10−01 | 9.36 × 10−02 | 4.31 × 10−01 | 1.07 × 10−01 | 1.67 × 10−01 |
| CCL5 | 8.38 × 10−02 | 2.80 × 10−01 | 9.98 × 10−02 | 3.18 × 10−01 | 3.17 × 10−01 | 2.20 × 10−01 |
| TNFSF10 | 3.27 × 10−01 | 1.49 × 10−01 | 1.76 × 10−01 | 3.88 × 10−01 | 2.06 × 10−02 | 5.12 × 10−02 |
| CSF1 | 1.79 × 10−01 | 4.38 × 10−01 | 2.57 × 10−01 | 1.23 × 10−01 | 2.20 × 10−01 | 9.77 × 10−02 |
| CCR4 | 4.57 × 10−01 | 1.79 × 10−01 | 2.66 × 10−01 | 4.10 × 10−01 | 1.59 × 10−01 | 1.48 × 10−01 |
| HLADRA | 1.61 × 10−01 | 5.11 × 10−01 | 2.73 × 10−01 | 4.78 × 10−01 | 1.21 × 10−01 | 2.16 × 10−01 |
| BAX | 9.26 × 10−01 | 5.98 × 10−02 | 2.86 × 10−01 | 9.97 × 10−01 | 2.79 × 10−02 | 1.94 × 10−01 |
| CD38 | 4.60 × 10−01 | 3.35 × 10−01 | 4.09 × 10−01 | 7.37 × 10−01 | 2.11 × 10−01 | 4.97 × 10−01 |
| SLAMF7 | 5.01 × 10−01 | 1.00 | 5.01 × 10−01 | 1.00 | 1.00 | 1.00 |
| GATA3 | 1.36 × 10−01 | 9.75 × 10−01 | 5.11 × 10−01 | 1.29 × 10−01 | 8.29 × 10−01 | 4.44 × 10−01 |
| PCNA | 8.92 × 10−01 | 3.40 × 10−01 | 5.52 × 10−01 | 8.19 × 10−01 | 1.00 | 1.00 |
| MMP9 | 6.35 × 10−01 | 1.00 | 6.35 × 10−01 | 1.00 | 1.00 | 1.00 |
| ENTPD1 | 6.50 × 10−01 | 1.00 | 6.50 × 10−01 | 2.97 × 10−02 | 1.00 | 3.10 × 10−02 |
| CCL4 | 6.79 × 10−01 | 6.22 × 10−01 | 7.55 × 10−01 | 1.00 | 1.00 | 1.00 |
| PRF1 | 9.51 × 10−01 | 4.12 × 10−01 | 7.67 × 10−01 | 7.96 × 10−01 | 1.00 | 1.00 |
| EOMES | 7.68 × 10−01 | 5.98 × 10−01 | 7.89 × 10−01 | 5.52 × 10−01 | 1.00 | 7.63 × 10−01 |
| IL6R | 6.31 × 10−01 | 7.39 × 10−01 | 7.98 × 10−01 | 2.22 × 10−01 | 5.60 × 10−01 | 5.96 × 10−01 |
| CCR5 | 4.52 × 10−01 | 9.28 × 10−01 | 8.09 × 10−01 | 1.33 × 10−01 | 5.54 × 10−01 | 3.07 × 10−01 |
| GZMA | 4.02 × 10−01 | 9.95 × 10−01 | 8.52 × 10−01 | 2.78 × 10−01 | 8.78 × 10−01 | 7.79 × 10−01 |
| CD8a | 9.58 × 10−01 | 1.00 | 9.58 × 10−01 | 6.68 × 10−01 | 1.00 | 9.00 × 10−01 |
| B3GAT1 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| CXCL13 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| IL12RbII | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| IL13 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| IL22 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| IL3 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| IL4 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| MKI67 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Gene Name | TMM | MAST | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gaussian | Binomial | Combine | Gaussian | Binomial | Combine | |||||||
| p-Value | FDR | p-Value | FDR | p-Value | FDR | p-Value | FDR | p-Value | FDR | p-Value | FDR | |
| TMSB15A | 2.14 × 10−12 | 5.31 × 10−10 | 1.60 × 10−55 | 3.33 × 10−51 | 1.57 × 10−55 | 3.27 × 10−51 | 4.50 × 10−09 | 4.66 × 10−07 | 1.42 × 10−44 | 3.01 × 10−40 | 1.35 × 10−44 | 2.86 × 10−40 |
| MEX3A | 2.01 × 10−10 | 2.66 × 10−08 | 1.55 × 10−50 | 1.62 × 10−46 | 1.34 × 10−50 | 1.39 × 10−46 | 3.73 × 10−08 | 2.79 × 10−06 | 1.57 × 10−42 | 1.67 × 10−38 | 1.57 × 10−42 | 1.67 × 10−38 |
| SPARCL1 | 2.31 × 10−15 | 1.42 × 10−12 | 1.53 × 10−49 | 1.07 × 10−45 | 1.29 × 10−49 | 8.94 × 10−46 | 1.22 × 10−13 | 6.46 × 10−11 | 7.91 × 10−38 | 5.60 × 10−34 | 7.32 × 10−38 | 5.18 × 10−34 |
| CLU | 3.86 × 10−14 | 1.75 × 10−11 | 7.64 × 10−43 | 3.98 × 10−39 | 7.54 × 10−43 | 3.93 × 10−39 | 3.24 × 10−12 | 1.11 × 10−09 | 1.36 × 10−33 | 5.78 × 10−30 | 1.26 × 10−33 | 5.23 × 10−30 |
| IL6ST | 2.78 × 10−06 | 8.37 × 10−05 | 5.91 × 10−42 | 2.46 × 10−38 | 5.24 × 10−42 | 2.19 × 10−38 | 5.82 × 10−04 | 7.40 × 10−03 | 3.49 × 10−33 | 1.06 × 10−29 | 3.17 × 10−33 | 9.61 × 10−30 |
| CRYAB | 1.90 × 10−13 | 6.51 × 10−11 | 4.47 × 10−39 | 1.55 × 10−35 | 4.06 × 10−39 | 1.41 × 10−35 | 2.62 × 10−11 | 6.78 × 10−09 | 1.66 × 10−34 | 8.82 × 10−31 | 1.43 × 10−34 | 7.60 × 10−31 |
| ALDOC | 1.30 × 10−16 | 9.72 × 10−14 | 2.84 × 10−36 | 8.46 × 10−33 | 2.28 × 10−36 | 6.79 × 10−33 | 2.50 × 10−14 | 1.87 × 10−11 | 2.93 × 10−29 | 5.66 × 10−26 | 2.65 × 10−29 | 5.11 × 10−26 |
| OSBPL1A | 3.47 × 10−20 | 6.03 × 10−17 | 1.09 × 10−35 | 2.76 × 10−32 | 9.29 × 10−36 | 2.35 × 10−32 | 4.44 × 10−20 | 1.35 × 10−16 | 1.71 × 10−33 | 6.07 × 10−30 | 1.48 × 10−33 | 5.23 × 10−30 |
| HTRA1 | 1.77 × 10−13 | 6.16 × 10−11 | 1.19 × 10−35 | 2.76 × 10−32 | 1.01 × 10−35 | 2.35 × 10−32 | 3.43 × 10−11 | 8.68 × 10−09 | 1.73 × 10−27 | 2.45 × 10−24 | 1.46 × 10−27 | 2.07 × 10−24 |
| PRNP | 6.70 × 10−24 | 3.50 × 10−20 | 2.33 × 10−35 | 4.87 × 10−32 | 2.24 × 10−35 | 4.66 × 10−32 | 5.61 × 10−19 | 1.32 × 10−15 | 4.92 × 10−30 | 1.16 × 10−26 | 4.04 × 10−30 | 9.53 × 10−27 |
| TSPYL2 | 1.46 × 10−19 | 2.03 × 10−16 | 8.68 × 10−35 | 1.65 × 10−31 | 8.53 × 10−35 | 1.62 × 10−31 | 8.81 × 10−19 | 1.87 × 10−15 | 6.39 × 10−29 | 1.13 × 10−25 | 6.17 × 10−29 | 1.09 × 10−25 |
| BHLHE41 | 3.97 × 10−12 | 9.01 × 10−10 | 1.08 × 10−34 | 1.88 × 10−31 | 9.52 × 10−35 | 1.66 × 10−31 | 3.60 × 10−08 | 2.70 × 10−06 | 6.76 × 10−28 | 1.03 × 10−24 | 6.45 × 10−28 | 9.79 × 10−25 |
| CD24 | 4.01 × 10−15 | 2.39 × 10−12 | 1.51 × 10−34 | 2.43 × 10−31 | 1.37 × 10−34 | 2.19 × 10−31 | 9.82 × 10−14 | 5.35 × 10−11 | 1.09 × 10−29 | 2.32 × 10−26 | 9.27 × 10−30 | 1.97 × 10−26 |
| NEUROD6 | 1.46 × 10−12 | 3.80 × 10−10 | 1.62 × 10−32 | 2.42 × 10−29 | 1.53 × 10−32 | 2.28 × 10−29 | 1.41 × 10−10 | 3.03 × 10−08 | 2.14 × 10−30 | 5.69 × 10−27 | 1.73 × 10−30 | 4.59 × 10−27 |
| ADD3 | 7.02 × 10−14 | 2.87 × 10−11 | 1.18 × 10−31 | 1.64 × 10−28 | 9.83 × 10−32 | 1.37 × 10−28 | 5.81 × 10−10 | 9.23 × 10−08 | 2.65 × 10−22 | 1.94 × 10−19 | 2.37 × 10−22 | 1.68 × 10−19 |
| BCL11A | 5.72 × 10−14 | 2.44 × 10−11 | 2.92 × 10−31 | 3.81 × 10−28 | 2.42 × 10−31 | 3.16 × 10−28 | 1.32 × 10−10 | 2.90 × 10−08 | 1.44 × 10−26 | 1.80 × 10−23 | 1.16 × 10−26 | 1.45 × 10−23 |
| SLC6A1 | 2.85 × 10−17 | 2.58 × 10−14 | 1.04 × 10−30 | 1.27 × 10−27 | 8.63 × 10−31 | 1.06 × 10−27 | 5.76 × 10−14 | 3.42 × 10−11 | 1.35 × 10−21 | 8.43 × 10−19 | 1.34 × 10−21 | 7.50 × 10−19 |
| NR3C1 | 5.03 × 10−07 | 1.93 × 10−05 | 5.15 × 10−30 | 5.66 × 10−27 | 4.30 × 10−30 | 4.89 × 10−27 | 1.20 × 10−09 | 1.68 × 10−07 | 3.01 × 10−27 | 4.00 × 10−24 | 3.06 × 10−27 | 4.06 × 10−24 |
| NEUROD2 | 2.34 × 10−06 | 7.27 × 10−05 | 4.56 × 10−30 | 5.29 × 10−27 | 4.45 × 10−30 | 4.89 × 10−27 | 7.12 × 10−06 | 2.22 × 10−04 | 3.74 × 10−28 | 6.11 × 10−25 | 3.68 × 10−28 | 6.01 × 10−25 |
| ALCAM | 5.90 × 10−15 | 3.33 × 10−12 | 6.98 × 10−30 | 7.28 × 10−27 | 5.98 × 10−30 | 6.24 × 10−27 | 1.80 × 10−16 | 2.25 × 10−13 | 2.25 × 10−23 | 1.99 × 10−20 | 2.13 × 10−23 | 1.81 × 10−20 |
References
- Ting, D.T.; Wittner, B.S.; Ligorio, M.; Vincent Jordan, N.; Shah, A.M.; Miyamoto, D.T.; Aceto, N.; Bersani, F.; Brannigan, B.W.; Xega, K.; et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014, 8, 1905–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef]
- Guo, G.; Huss, M.; Tong, G.Q.; Wang, C.; Li Sun, L.; Clarke, N.D.; Robson, P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev. Cell 2010, 18, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacher, R.; Kendziorski, C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biol. 2016, 17, 63. [Google Scholar] [CrossRef] [Green Version]
- McDavid, A.; Finak, G.; Chattopadyay, P.K.; Dominguez, M.; Lamoreaux, L.; Ma, S.S.; Roederer, M.; Gottardo, R. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics 2013, 29, 461–467. [Google Scholar] [CrossRef]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Hayden Gephart, M.G.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef] [Green Version]
- Duan, N.; Manning, W.G.; Morris, C.N.; Newhouse, J.P. A comparison of alternative models for the demand for medical care. J. Bus. Econ. Stat. 1983, 1, 115–126. [Google Scholar]
- Duan, N.; Manning, W.G.; Morris, C.N.; Newhouse, J.P. Choosing between the sample-selection model and the multi-part model. J. Bus. Econ. Stat. 1984, 2, 283–289. [Google Scholar]
- Min, Y.; Agresti, A. Modeling nonnegative data with clumping at zero: A survey. J. Iran. Stat. Soc. 2002, 1, 7–33. [Google Scholar]
- Olsen, M.K.; Schafer, J.L. A two-part random-effects model for semicontinuous longitudinal data. J. Am. Stat. Assoc. 2001, 96, 730–745. [Google Scholar] [CrossRef]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, D.A.; Skaug, H.J.; Ancheta, J.; Ianelli, J.; Magnusson, A.; Maunder, M.N.; Nielsen, A.; Sibert, J. AD Model Builder: Using automatic differentiation for statistical inference of highly parameterized complex nonlinear models. Optim. Methods Softw. 2012, 27, 233–249. [Google Scholar] [CrossRef] [Green Version]
- Skaug, H.J.; Fournier, D.A. Automatic approximation of the marginal likelihood in non-gaussian hierarchical models. Comput. Stat. Data Anal. 2006, 51, 699–709. [Google Scholar] [CrossRef]
- Pinheiro, J.; Bates, D. Mixed-Effects Models in S and S-PLUS; Springer Science & Business Media: New York, NY, USA, 2006. [Google Scholar]
- Liu, L.; Strawderman, R.L.; Cowen, M.E.; Shih, Y.C. A flexible two-part random effects model for correlated medical costs. J. Health Econ. 2010, 29, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Halekoh, U.; Højsgaard, S. A kenward-roger approximation and parametric bootstrap methods for tests in linear mixed models–the R package pbkrtest. J. Stat. Softw. 2014, 59, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Efron, B.; Tibshirani, R.J. An Introduction to the Bootstrap; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Davison, A.C.; Hinkley, D.V. Bootstrap Methods and Their Application; Cambridge University Press: Cambridge, UK, 1997; Volume 1. [Google Scholar]
- Cai, T.; Lin, X.; Carroll, R.J. Identifying genetic marker sets associated with phenotypes via an efficient adaptive score test. Biostatistics 2012, 13, 776–790. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.T.; Lin, X. Gene set analysis using variance component tests. BMC Bioinform. 2013, 14, 210. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Lin, X.; Ghosh, D. Semiparametric regression of multidimensional genetic pathway data: Least-squares kernel machines and linear mixed models. Biometrics 2007, 63, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, M.H.; Chattopadhyay, P.K.; Ma, S.; Lamoreaux, L.; McDavid, A.; Finak, G.; Gottardo, R.; Koup, R.A.; Roederer, M. Highly multiplexed quantitation of gene expression on single cells. J. Immunol. Methods 2013, 391, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squair, J.W.; Gautier, M.; Kathe, C.; Anderson, M.A.; James, N.D.; Hutson, T.H.; Hudelle, R.; Qaiser, T.; Matson, K.J.E.; Barraud, Q.; et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021, 12, 5692. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Cano-Gamez, E.; Soskic, B.; Roumeliotis, T.I.; So, E.; Smyth, D.J.; Baldrighi, M.; Wille, D.; Nakic, N.; Esparza-Gordillo, J.; Larminie, C.G.C.; et al. Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4(+) T cells to cytokines. Nat. Commun. 2020, 11, 1801. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Society Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Hicks, S.C.; Teng, M.; Irizarry, R.A. On the widespread and critical impact of systematic bias and batch effects in single-cell RNA-Seq data. bioRxiv 2015, 10, 025528. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Lee, J.-H.; Kang, H.; Jiang, H. A Two-Part Mixed Model for Differential Expression Analysis in Single-Cell High-Throughput Gene Expression Data. Genes 2022, 13, 377. https://doi.org/10.3390/genes13020377
Shi Y, Lee J-H, Kang H, Jiang H. A Two-Part Mixed Model for Differential Expression Analysis in Single-Cell High-Throughput Gene Expression Data. Genes. 2022; 13(2):377. https://doi.org/10.3390/genes13020377
Chicago/Turabian StyleShi, Yang, Ji-Hyun Lee, Huining Kang, and Hui Jiang. 2022. "A Two-Part Mixed Model for Differential Expression Analysis in Single-Cell High-Throughput Gene Expression Data" Genes 13, no. 2: 377. https://doi.org/10.3390/genes13020377
APA StyleShi, Y., Lee, J.-H., Kang, H., & Jiang, H. (2022). A Two-Part Mixed Model for Differential Expression Analysis in Single-Cell High-Throughput Gene Expression Data. Genes, 13(2), 377. https://doi.org/10.3390/genes13020377