
Identification of Genomic Regions Associated with Agronomic and Disease Resistance Traits in a Large Set of Multiple DH Populations

 , ,
, ,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Field Trials
2.2. Mass Production and Artificial Inoculation of MLN Viruses
2.3. Phenotyping and Data Analysis
2.4. Genotypic Data Analyses
2.5. Population Structure and GWAS
2.6. Genomic Predictions
3. Results
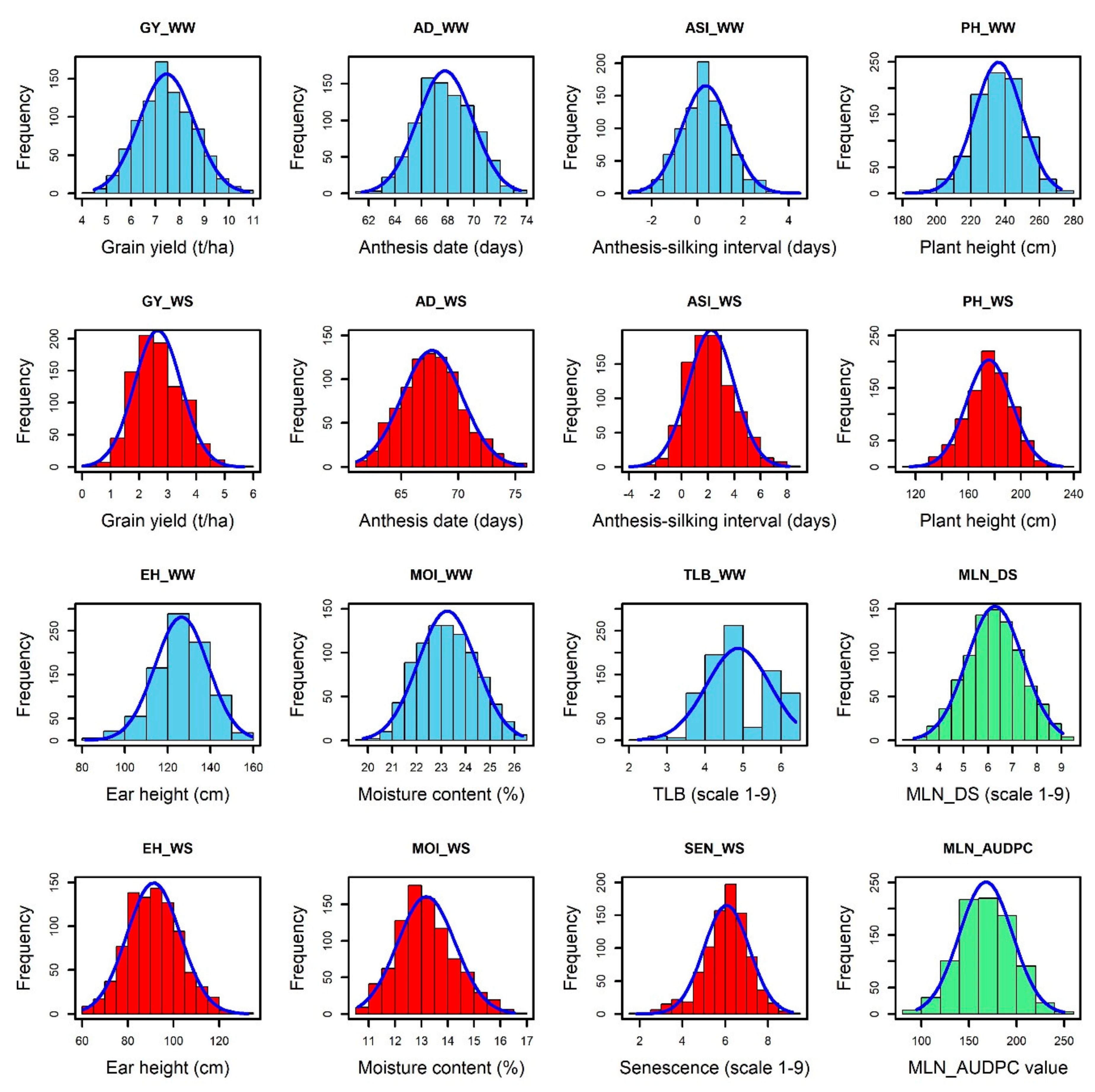
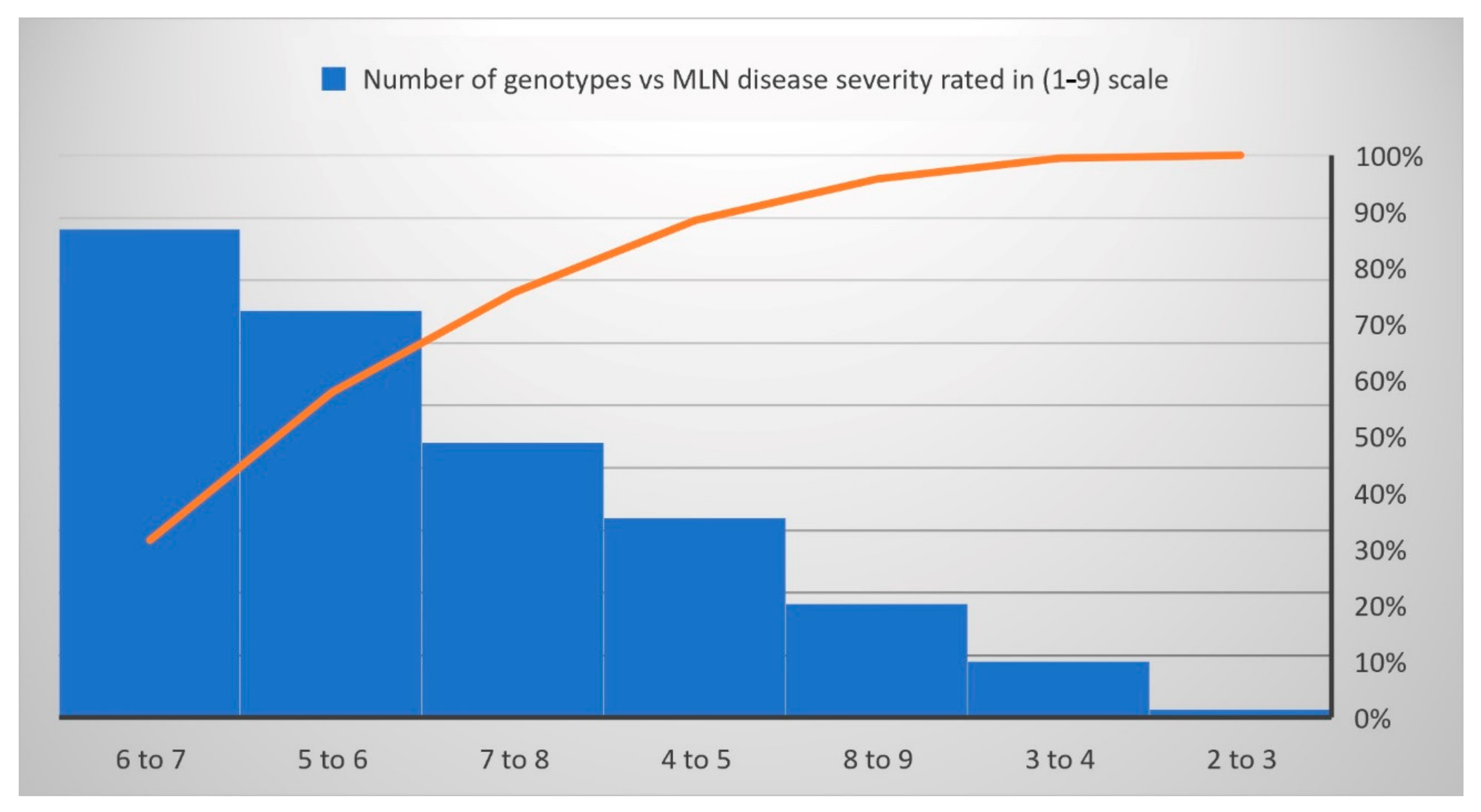
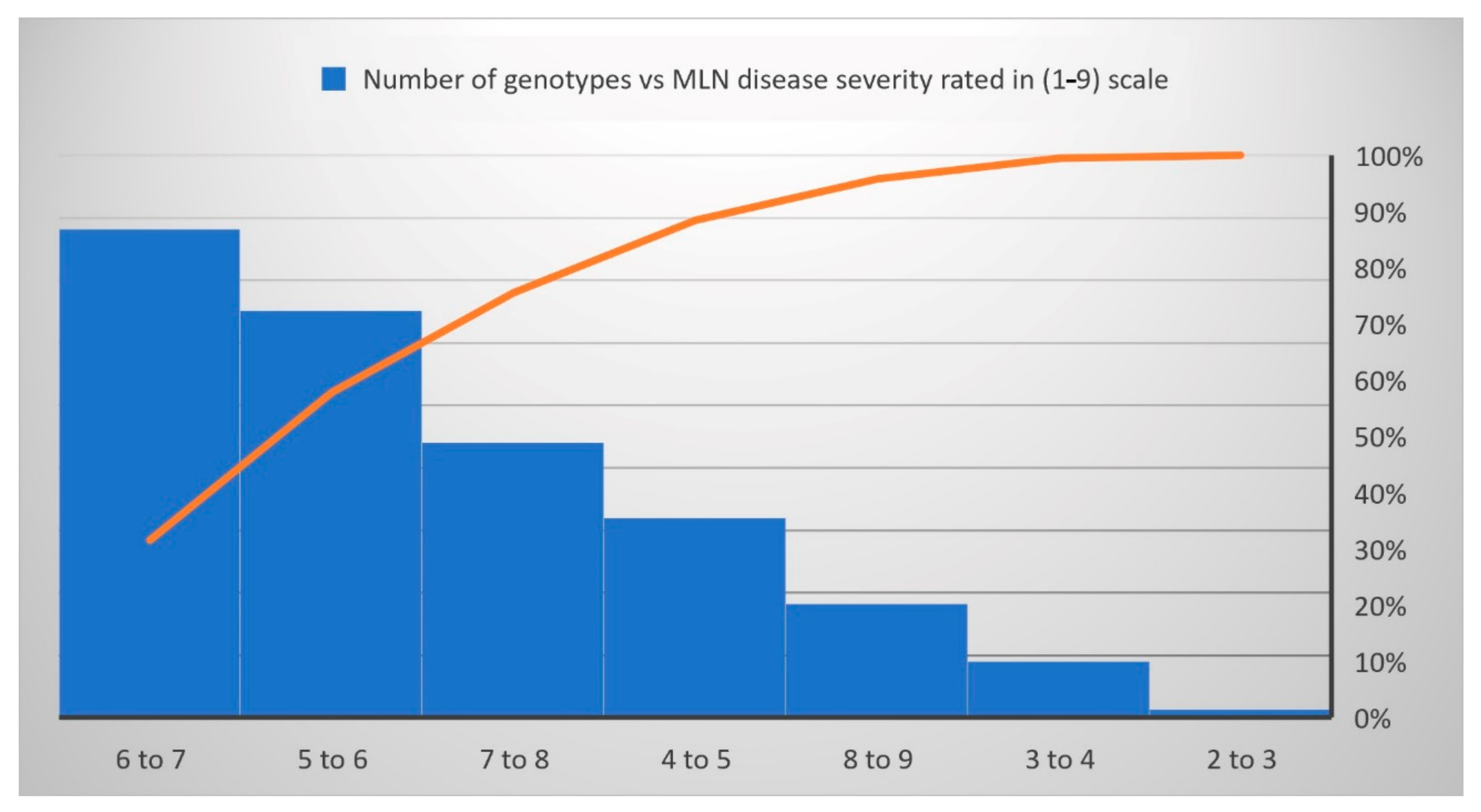
3.1. Phenotypic Variations and Correlations
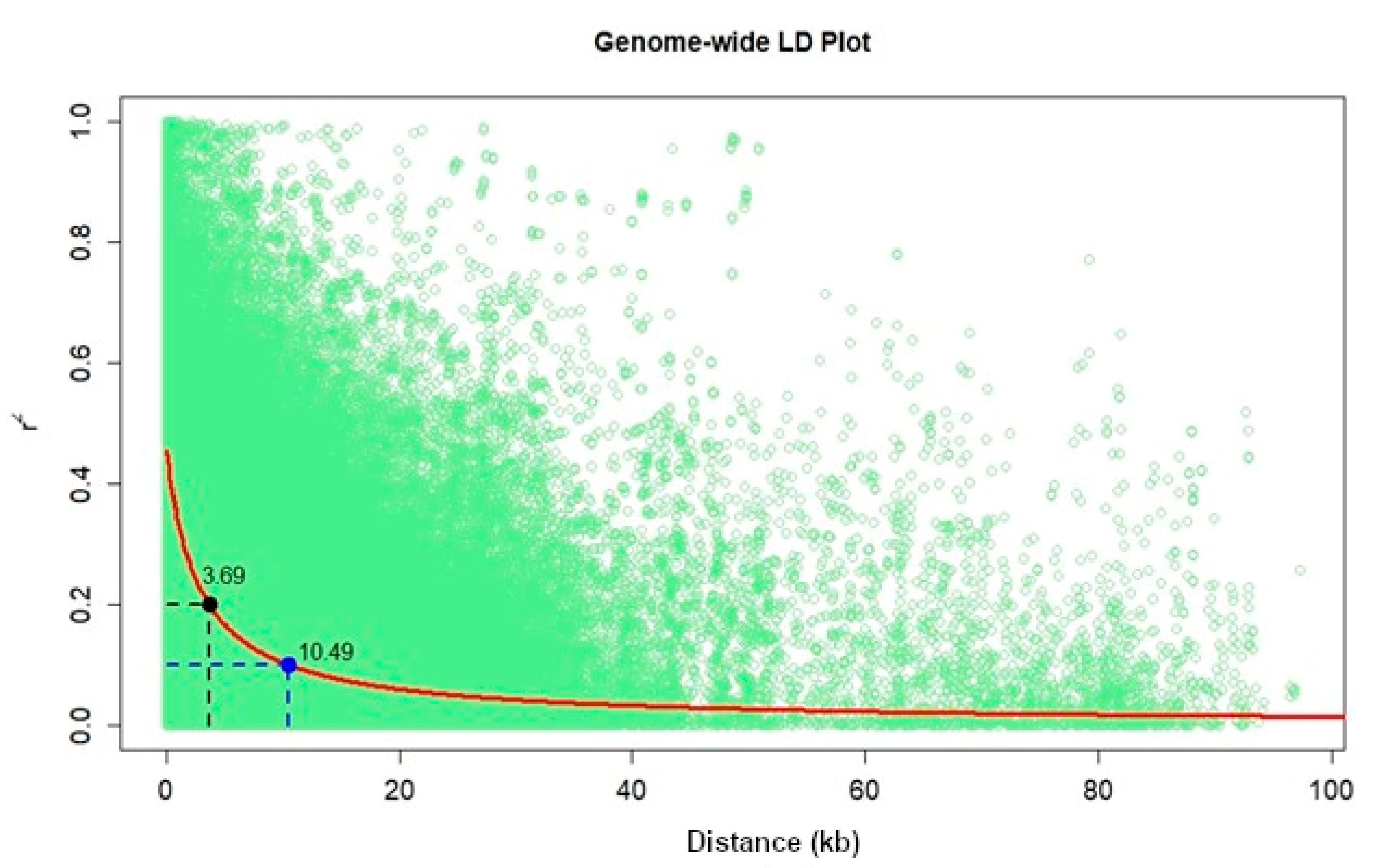
3.2. Genetic Relationship, Population Structure, and Linkage Disequilibrium
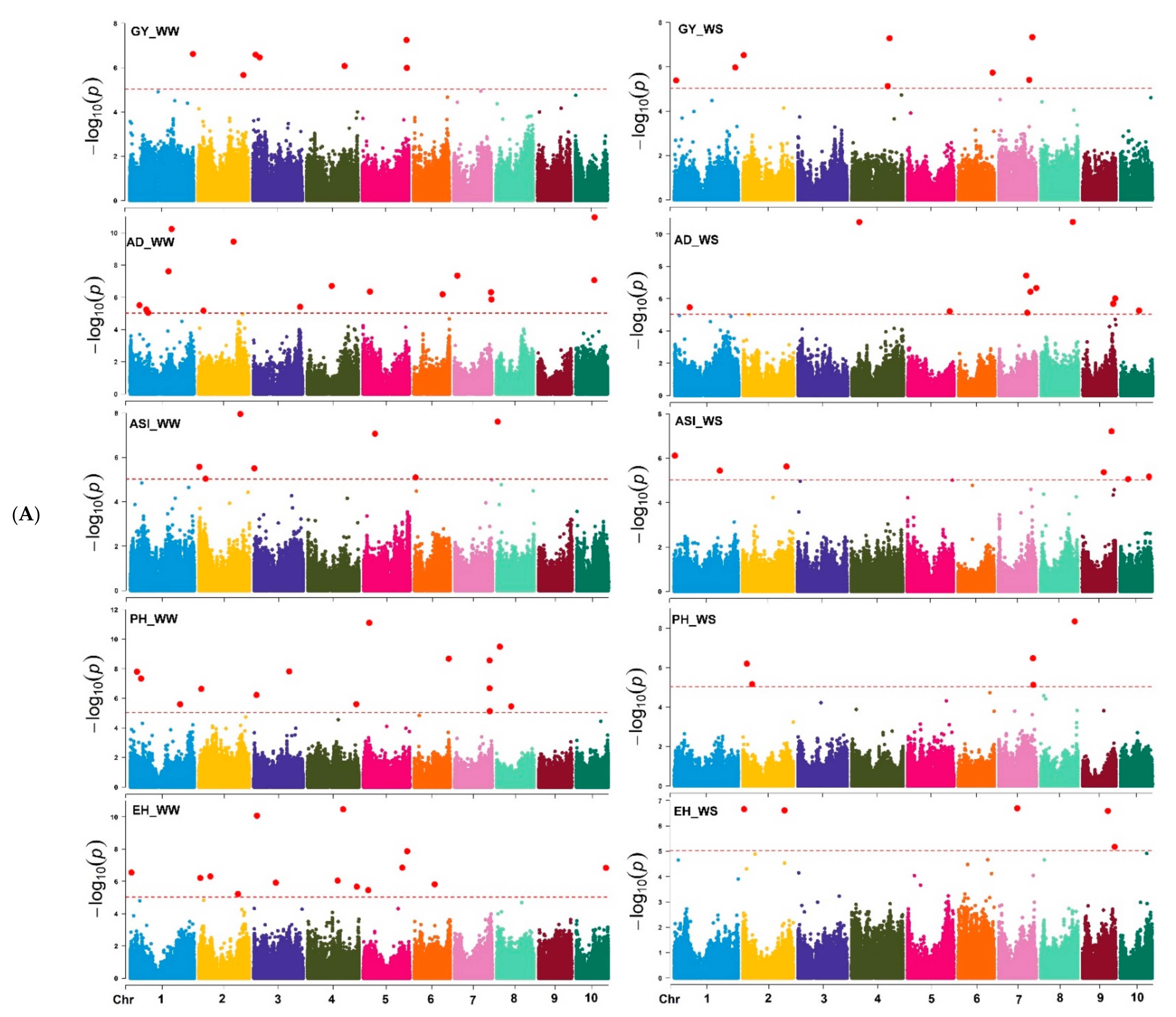
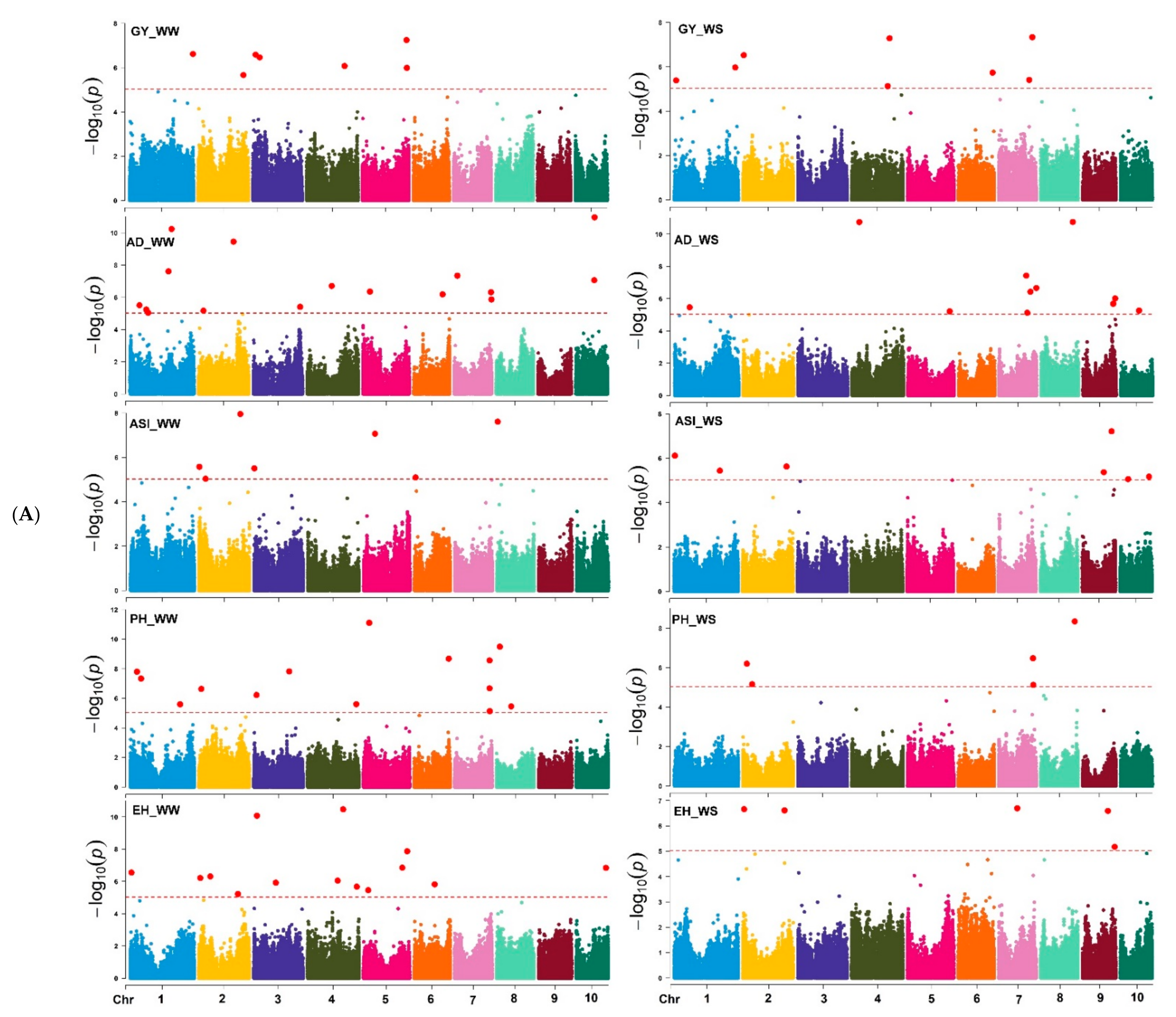
3.3. GWAS Results
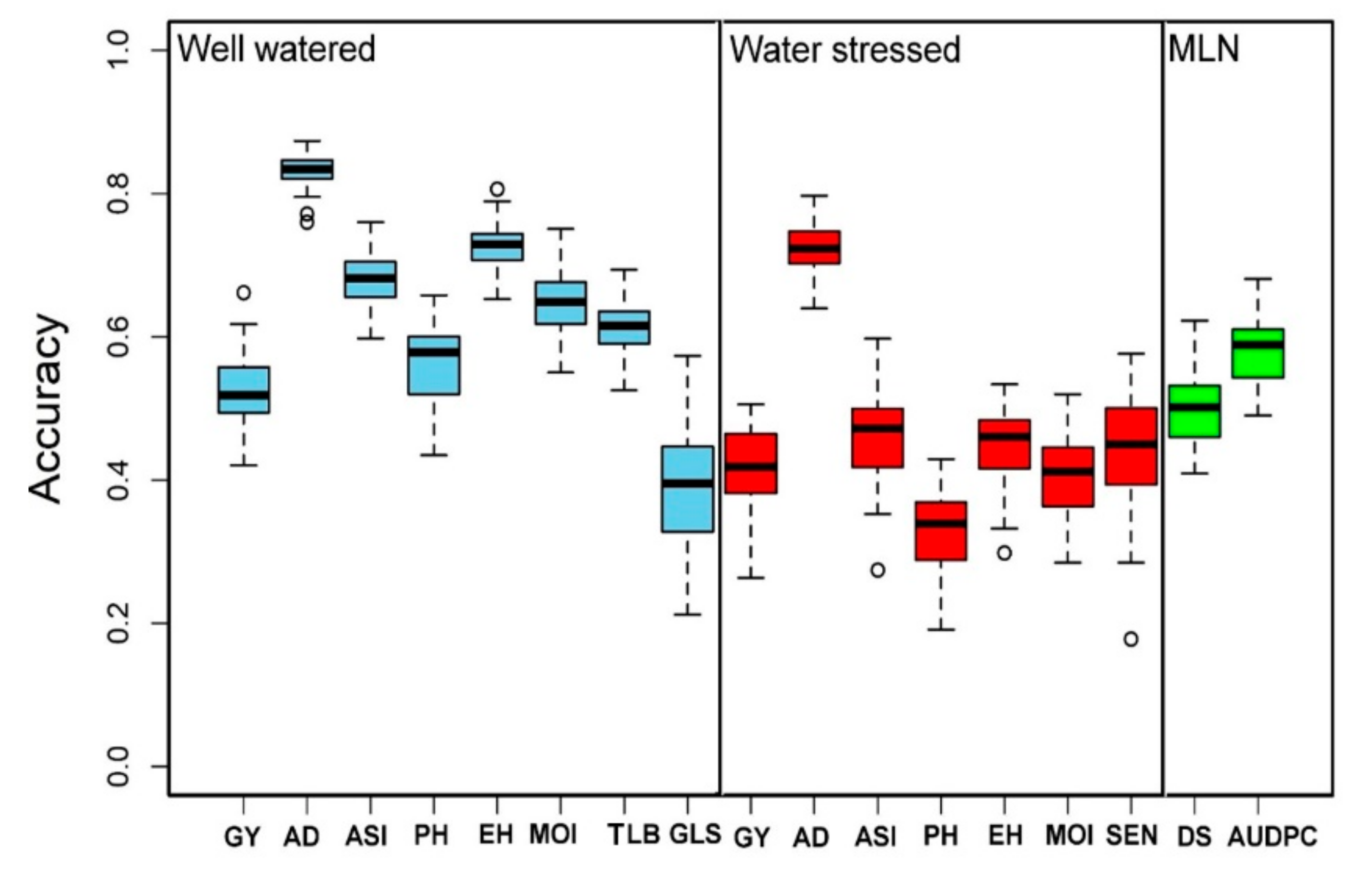
3.4. Genomic Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santpoort, R. THE drivers of maize area expansion in sub-Saharan Africa. How policies to boost maize production overlook the interests of smallholder farmers. Land 2020, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Semagn, K.; Beyene, Y.; Warburton, M.L. Meta-analyses of QTL for grain yield and anthesis silking interval in 18 maize populations evaluated under water-stressed and well-watered environments. BMC Genom. 2013, 14, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangai, A.W.; Reibaugh, M.G.; Kiyua, Z.M. Maize Lethal Necrosis in Kenya. Plant Dis. 2012, 96, 1582. [Google Scholar] [CrossRef]
- Adams, I.P.; Harju, V.; Hodges, T. First report of maize lethal necrosis disease in Rwanda. New Dis. Rep. 2014, 29, 22. [Google Scholar] [CrossRef] [Green Version]
- Mahuku, G.; Lockhart, B.E.; Wanjala, B.; Jones, M.W.; Kimunye, J.N.; Stewart, L.R.; Cassone, B.J.; Sevgan, S.; Nyasani, J.O.; Kusia, E.; et al. Maize lethal necrosis (MLN), an emerging threat to maize-based food security in sub-Saharan Africa. Phytopathology 2015, 105, 956–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Zhang, J.; Wang, Q.; Meng, C.; Hong, J.; Zhou, X. Characterization of Maize Chlorotic Mottle Virus Associated with Maize Lethal Necrosis Disease in China. J. Phytopathol. 2011, 159, 191–193. [Google Scholar] [CrossRef]
- Redinbaugh, M.G.; Stewart, L.R. Maize lethal necrosis: An emerging, synergistic viral disease. Annu. Rev. Virol. 2018, 5, 301–322. [Google Scholar] [CrossRef]
- Nyasani, J.O.; Meyhöfer, R.; Subramanian, S.; Poehling, H.M. Effect of intercrops on thrips species composition and population abundance on French beans in Kenya. Entomol. Exp. Appl. 2012, 142, 236–246. [Google Scholar] [CrossRef]
- Jensen, S.G.; Wysong, D.; Ball, E.; Higley, P. Seed transmission of MCMV. Plant Dis. 1991, 75, 497–498. [Google Scholar] [CrossRef]
- Niblett, C.L.; Claflin, L.E. Corn lethal necrosis a new virus disease of corn in Kansas. Plant Dis. Rep. 1978, 62, 15–19. [Google Scholar]
- Zhao, M.; Ho, H.; Wu, Y.; He, Y.; Li, M. Western Flower Thrips (Frankliniella occidentalis) Transmits Maize Chlorotic Mottle Virus. J. Phytopathol. 2014, 162, 532–536. [Google Scholar] [CrossRef]
- Gowda, M.; Beyene, Y.; Makumbi, D.; Semagn, K.; Olsen, M.S.; Bright, J.M.; Das, B.; Mugo, S.; Suresh, L.M.; Prasanna, B.M. Discovery and validation of genomic regions associated with resistance to maize lethal necrosis in four biparental populations. Mol. Breed. 2018, 38, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awata, O.L.; Ifei, B.E.; Tongoona, P.; Danquah, E.; Jumbo, M.B.; Gowda, M.; Marchelo-D’ragga, P.W.; Sitonik, C.A.; Suresh, L.M. Maize lethal necrosis and the molecular basis of variability in concentrations of the causal viruses in co-infected maize plant. J. Gen. Mol. Virol. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Yan, G.; Liu, H.; Wang, H.; Lu, Z.; Wang, Y.; Mullan, D.; Hamblin, J.; Liu, C. Accelerated generation of selfed pure line plants for gene identification and crop breeding. Front. Plant Sci. 2017, 8, 1786. [Google Scholar] [CrossRef] [Green Version]
- Mayor, P.J.; Bernardo, R. Genomewide selection and marker-assisted recurrent selection in doubled haploid versus F2 populations. Crop Sci. 2009, 49, 1719–1725. [Google Scholar] [CrossRef]
- Bänziger, M.; Betrán, F.J.; Lafitte, H.R. Efficiency of high-nitroGen. selection environments for improving maize for low-nitroGen. target environments. Crop Sci. 1997, 37, 1103–1109. [Google Scholar] [CrossRef]
- Bänziger, M.; Setimela, P.S.; Hodson, D.; Vivek, B. Breeding for improved abiotic stress tolerance in maize adapted to southern Africa. Agric. Water Manag. 2006, 80, 212–224. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Mol. Plant. 2017, 10, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Buckler, E.; Ersoz, E.S.; Yu, J.; Buckler, E.S. Applications of Linkage Disequilibrium and Association Mapping in Maize Ap-plications of Linkage Disequilibrium and Association Mapping in Maize. In Molecular Genetic Approaches to Maize Improvement; Springer: Dordrecht, The Netherlands, 2009; Volume 63, pp. 173–195. [Google Scholar] [CrossRef]
- Ching, A.; Caldwell, K.S.; Jung, M.; Dolan, M.; Smith, O.S.; Tingey, S.; Morgante, M.; Rafalski, A.J. SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet. 2002, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Collard, B.Y.; Jahufer, M.Z.; Brouwer, J.B.; Pang, E.C. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar] [CrossRef]
- Soller, M.; Beckmann, J.S. Genetic polymorphism in varietal identification and genetic improvement. Theor. Appl. Genet. 1983, 67, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E.; Kemper, K.E.; MacLeod, I.M.; Chamberlain, A.J.; Hayes, B.J. Genetics of complex traits: Prediction of phenotype, identification of causal polymorphisms and genetic architecture. Proc. R. Soc. B 2016, 283, 20160569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Cairns, J.E.; Babu, R.; Gowda, M.; Makumbi, D.; Magorokosho, C.; Zhang, A.; Liu, Y.; Wang, N.; Hao, Z.; et al. Genome-wide association mapping and genomic prediction analyses reveal the genetic architecture of grain yield and flowering time under drought and heat stress conditions in maize. Front. Plant Sci. 2019, 9, 1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Z.; Li, X.; Liu, X. Meta-analysis of constitutive and adaptive QTL for drought tolerance in maize. Euphytica 2010, 174, 165–177. [Google Scholar] [CrossRef]
- Xu, J.; Yuan, Y.; Xu, Y.; Zhang, G.; Guo, X.; Wu, F.; Wang, Q.; Rong, T.; Pan, G.; Cao, M.; et al. Identification of candidate genes for drought tolerance by whole-genome resequencing in maize. BMC Plant Biol. 2014, 14, 83. [Google Scholar] [CrossRef] [Green Version]
- Agrama, H.; Moussa, M.E. Mapping QTLs in breeding for drought tolerance in maize (Zea mays L.). Euphytica 1996, 91, 89–97. [Google Scholar] [CrossRef]
- Yan, W.; Jackson, A.; Jia, M.; Zhou, W.; Xiong, H.; Bryant, R. Association Mapping of Four Important Traits Using the USDA Rice Mini-Core Collection. In Rice-Germplasm Genetics Improvement; Intech Open: London, UK, 2014; pp. 105–142. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, P.A.M.; Fritsche-Neto, R.; Andrade, M.C.; Petroli, C.D.; Burgueño, J.; Galli, G.; Willcox, M.C.; Sonder, K.; Vidal-Martínez, V.A.; Sifuentes-Ibarra, E.; et al. Introgression of maize diversity for drought tolerance subtropical maize landraces as source of new positive variants. In Front. Plant Sci.; 2021; Volume 12, p. 691211. [Google Scholar] [CrossRef]
- Olsen, M.; Yao, N.; Tadesse, B.; Das, B.; Gowda, M.; Semagn, K.; Jumbo, M.; Killian, A. Mapping Genomic Regions Associated with Maize Lethal Necrosis (MLN) Using QTL-Seq; Divers Arrays Technol Pty Ltd. International Livestock Research Institute: Nairobi, Kenya, 2016; pp. 1–4. [Google Scholar] [CrossRef]
- Gowda, M.; Das, B.; Makumbi, D.; Babu, R.; Semagn, K.; Mahuku, G.; Olsen, M.S.; Bright, J.M.; Beyene, Y.; Prasanna, B.M. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 2015, 128, 1957–1968. [Google Scholar] [CrossRef] [Green Version]
- Boddupalli, P.; Suresh, L.; Mwatuni, F.; Beyene, Y.; Makumbi, D.; Gowda, M.; Olsen, M.; Hodson, D.; Worku, M.; Mezzalama, M.; et al. Maize lethal necrosis (MLN): Efforts toward containing the spread and impact of a devastating transboundary disease in sub-Saharan Africa. Virus Res. 2020, 282, 197943. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Poland, J.A.; Rife, T.W. Genotyping-by-Sequencing for Plant Breeding and Genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.-L.; McCouch, S.R. Genomic Selection and Association Mapping in Rice (Oryza sativa): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Gowda, M.; Liu, W.; Würschum, T.; Maurer, H.P.; Longin, F.H.; Ranc, N.; Reif, J.C. Accuracy of genomic selection in European maize elite breeding populations. Theor. Appl. Genet. 2012, 124, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Nyaga, C.; Gowda, M.; Beyene, Y.; Murithi, W.T.; Burgueno, J.; Toledo, F.; Makumbi, D.; Olsen, M.S.; Das, B.; LM, S.; et al. Hybrid breeding for MLN resistance: Heterosis, combining ability, and hybrid prediction. Plants 2020, 9, 468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitonik, C.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Makumbi, D.; Oliver, K.; Das, B.; Bright, J.M.; Mugo, S.; Crossa, J.; et al. Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor. Appl. Genet. 2019, 132, 2381–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarado, G.; López, M.; Vargas, M.; Pacheco, A.; Rodríguez, F. USER’S MANUAL META-R (Multi-Environment Trial Analysis with R); CIMMYT: El Batan, Mexico, 2015; pp. 1–34. [Google Scholar]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CIMMYT. Laboratory Protocols: CIMMYT Applied Molecular Genetics Laboratory; CIMMYT: El Batan, Mexico, 2005; Available online: http://repository.cimmyt.org/xmlui/bitstream/handle/10883/1333/91195.pdf (accessed on 11 November 2020).
- Cao, S.; Loladze, A.; Yuan, Y.; Wu, Y.; Zhang, A.; Chen, J.; Huestis, G.; Cao, J.; Chaikam, V.; Olsen, M.; et al. Genome-Wide Analysis of Tar Spot Complex Resistance in Maize Using Genotyping-by-Sequencing SNPs and Whole-Genome Prediction. Plant Genome 2017, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef]
- Hill, W.G.; Weir, B.S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- Remington, D.L.; Thornsberry, J.M.; Matsuoka, Y.; Wilson, L.M.; Whitt, S.R.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2016. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, D.A.; Von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.; Luo, J.; Qi, C.; Ruan, Y.; Li, J.; Zhang, A.; Yang, X.; He, Y. Genome-wide association study (GWAS) reveals the genetic architecture of four husk traits in maize. BMC Genom. 2016, 17, 946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyaga, C.; Gowda, M.; Beyene, Y.; Muriithi, W.T. Genome-Wide Analyses and Prediction of Resistance to MLN in Large Tropical Maize Germplasm. Genes 2020, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Kibe, M.; Nyaga, C.; Nair, S.K.; Beyene, Y.; Das, B.; LM, S.L.; Bright, J.M.; Makumbi, D.; Kinyua, J.; Olsen, M.S.; et al. Combination of linkage mapping, gwas, and gp to dissect the genetic basis of common rust resistance in tropical maize germplasm. Int. J. Mol. Sci. 2020, 21, 6518. [Google Scholar] [CrossRef]
- De Groote, H.; Oloo, F.; Tongruksawattana, S.; Das, B. Community-survey based assessment of the geographic distribution and impact of maize lethal necrosis (MLN) disease in Kenya. Crop Prot. 2016, 82, 30–35. [Google Scholar] [CrossRef]
- Beyene, Y.; Gowda, M.; Suresh, L.M.; Mugo, S.; Olsen, M.; Oikeh, S.O.; Juma, C.; Tarekegne, A.; Prasanna, B.M. Genetic analysis of tropical maize inbred lines for resistance to maize lethal necrosis disease. Euphytica 2017, 213, 224. [Google Scholar] [CrossRef] [Green Version]
- Myles, S.; Peiffer, J.; Brown, P.J.; Ersoz, E.S.; Zhang, Z.; Costich, D.E.; Buckler, E. Association mapping: Critical considerations shift from genotyping to experimental design. Plant Cell. 2009, 21, 2194–2202. [Google Scholar] [CrossRef] [Green Version]
- Rashid, Z.; Singh, P.K.; Vemuri, H.; Zaidi, P.H.; Prasanna, B.M.; Nair, S.K. Genome-wide association study in Asia-adapted tropical maize reveals novel and explored genomic regions for sorghum downy mildew resistance. Sci. Rep. 2018, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yan, J.; Guimarães, C.T.; Taba, S.; Hao, Z.; Gao, S.; Chen, S.; Li, J.; Zhang, S.; Vivek, B.S.; et al. Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor. Appl. Genet. 2009, 120, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Ertiro, B.T.; Labuschagne, M.; Olsen, M.; Das, B.; Prasanna, B.M.; Gowda, M. Genetic Dissection of NitroGen. Use Efficiency in Tropical Maize Through Genome-Wide Association and Genomic Prediction. Plant Sci. 2020, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, H.; Beyene, Y.; Semagn, K.; Liu, Y.; Cao, S.; Cui, Z.; Ruan, Y.; Burgueño, J.; Vicente, F.S.; et al. Effect of trait heritability, training population size and marker density on genomic prediction accuracy estimation in 22 bi-parental tropical maize populations. Front. Plant Sci. 2017, 8, 1916. [Google Scholar] [CrossRef]
- Zhang, X.; Pérez-Rodríguez, P.; Semagn, K.; Beyene, Y.; Babu, R.; A López-Cruz, M.; Vicente, F.S.; Olsen, M.; Buckler, E.; Jannink, J.-L.; et al. Genomic prediction in biparental tropical maize populations in water-stressed and well-watered environments using low-density and GBS SNPs. Heredity 2015, 114, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Sani, H.M.; Hamzeh-Mivehroud, M.; Silva, A.P.; Walshe, J.L.; Mohammadi, S.A.; Rahbar-Shahrouziasl, M.; Abbasi, M.; Jamshidi, O.; Low, J.K.; Dastmalchi, S.; et al. Expression, purification and DNA-binding properties of zinc finger domains of DOF proteins from Arabidopsis thaliana. BioImpacts 2018, 8, 167–176. [Google Scholar] [CrossRef]
- Islam, M.R.; Kim, H.; Kang, S.-W.; Kim, J.-S.; Jeong, Y.-M.; Hwang, H.-J.; Lee, S.-Y.; Woo, J.-C.; Kim, S.-G. Functional characterization of a gene encoding a dual domain for uridine kinase and uracil phosphoribosyltransferase in Arabidopsis thaliana. Plant Mol. Biol. 2007, 63, 465–477. [Google Scholar] [CrossRef]
- Huang, Y.; Lemieux, M.J.; Song, J.; Auer, M.; Wang, D.N. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 2003, 301, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Cosio, C.; Dunand, C. Specific functions of individual class III peroxdase genes. J. Exp. Bot. 2009, 60, 391–408. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, Y.; Liang, Y.; Zhou, D.; Li, S.; Lin, S.; Dong, H.; Huang, L. The function of histone lysine methylation related SET domain group proteins in plants. Protein Sci. 2020, 29, 1120–1137. [Google Scholar] [CrossRef]
- Esch, L.; Schaffrath, U. An update on jacalin-like lectins and their role in plant defense. Int. J. Mol. Sci. 2017, 18, 1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svennerstam, H.; Ganeteg, U.; Bellini, C.; Näsholm, T. Comprehensive screening of Arabidopsis mutants suggests the lysine histidine transporter 1 to be involved in plant uptake of amino acids. Plant Physiol. 2007, 143, 1853–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, T.; Kong, Z.; Ho, C.-M.K.; Zeng, C.J.T.; Horio, T.; Fong, S.; Vuong, T.; Lee, Y.-R.J.; Liu, B. Characterization of the Arabidopsis augmin complex uncovers its critical function in the assembly of the acentrosomal spindle and phragmoplast microtubule arrays. Plant Cell. 2012, 24, 1494–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallister, C.; Siracusa, M.C.; Shirazi, F.; Chalkia, D.; Nikolaidis, N. Functional diversification and specialization of cytosolic 70-kDa heat shock proteins. Sci. Rep. 2015, 5, srep09363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohorn, B.D.; Kohorn, S.L. The cell wall-associated kinases, WAKs, as pectin receptors. Front. Plant Sci. 2012, 3, 88. [Google Scholar] [CrossRef] [Green Version]
- Zhong, R.; Cui, D.; Phillips, D.R.; Ye, Z.H. A Novel Rice Xylosyltransferase Catalyzes the Addition of 2-O-Xylosyl Side Chains onto the Xylan Backbone. Plant Cell Physiol. 2018, 59, 554–565. [Google Scholar] [CrossRef]
- Mudd, E.A.; Sullivan, S.; Gisby, M.F.; Mironov, A.; Kwon, C.S.; Chung, W.-I.; Day, A. A 125 kDa RNase E/G-like protein is present in plastids and is essential for chloroplast development and autotrophic growth in Arabidopsis. J. Exp. Bot. 2008, 59, 2597–2610. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; He, B.; Liu, X.; Ma, X.; Liu, Y.; Yao, H.; Zhang, P.; Yin, J.; Wei, X.; Koh, H.; et al. Pyrophosphate-fructose 6-phosphate 1-phosphotransferase (PFP1) regulates starch biosynthesis and seed development via heterotetramer formation in rice (Oryza sativa L.). Plant Biotechnol. J. 2020, 18, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Akoh, C.C.; Lee, G.C.; Liaw, Y.C.; Huang, T.H.; Shaw, J.F. GDSL family of serine esterases/lipases. Prog. Lipid Res. 2004, 43, 534–552. [Google Scholar] [CrossRef]
- Rédei, G.P. Major Facilitator Superfamily (MFS). In Encyclopedia of Genetics, Genomics, Proteomics, and Informatics; Springer: Dordrecht, The Netherlands, 2008; Volume 62, p. 1142. [Google Scholar] [CrossRef]
- Chai, M.F.; Chen, Q.J.; An, R.; Chen, Y.M.; Chen, J.; Wang, X.C. NADK2, an Arabidopsis chloroplastic NAD kinase, plays a vital role in both chlorophyll synthesis and chloroplast protection. Plant Mol. Biol. 2005, 59, 553–564. [Google Scholar] [CrossRef]
- Richmond, G.S.; Smith, T.K. Phospholipases A 1. Int. J. Mol. Sci. 2011, 12, 588–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anil, K.S.; Hima, K.P.; Shravan, K.G.; Mohanalatha, C.; Kavi, K.P. Osmotin: A plant sentinel and a possible agonist of mammalian adiponectin. Front. Plant Sci. 2015, 6, 163. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Lai, Y.; Lv, L.; Ji, M.; Han, K.; Yan, D.; Lu, Y.; Peng, J.; Rao, S.; Yan, F.; et al. Fasciclin-like arabinogalactan gene family in Nicotiana benthamiana: Genome-wide identification, classification and expression in response to pathogens. BMC Plant Biol. 2020, 20, 305. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Kandianis, C.B.; E Harjes, C.; Bai, L.; Kim, E.-H.; Yang, X.; Skinner, D.J.; Fu, Z.; Mitchell, S.; Li, Q.; et al. Rare genetic variation at Zea mays crtRB1 increases Β-carotene in maize grain. Nat. Genet. 2010, 42, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirunavukkarasu, N.; Hossain, F.; Arora, K.; Sharma, R.; Shiriga, K.; Mittal, S.; Mohan, S.; Namratha, P.M.; Dogga, S.; Rani, T.S.; et al. Functional mechanisms of drought tolerance in subtropical maize (Zea mays L.) identified using genome-wide association mapping. BMC Genom. 2014, 15, 1182. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wang, Z.; Liang, X.; Weng, J.; Lv, X.; Zhang, D.; Yang, J.; Yong, H.; Li, M.; Li, F.; et al. Identification of loci contributing to maize drought tolerance in a genome-wide association study. Euphytica 2016, 210, 165–179. [Google Scholar] [CrossRef]
- Suwarno, W.B.; Pixley, K.V.; Palacios-Rojas, N.; Kaeppler, S.M.; Babu, R. Genome-wide association analysis reveals new targets for carotenoid biofortification in maize. Theor. Appl. Genet. 2015, 128, 851–864. [Google Scholar] [CrossRef] [Green Version]
- Beyene, Y.; Gowda, M.; Pérez-Rodríguez, P.; Olsen, M.; Robbins, K.R.; Burgueño, J.; Prasanna, B.M.; Crossa, J. Application of Genomic Selection at the Early Stage of Breeding Pipeline in Tropical Maize. Front. Plant Sci. 2021, 12, 685488. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Hickey, J.; Burgueño, J.; Ornella, L.; Cerón-Rojas, J.J.; Zhang, X.; Dreisigacker, S.; Babu, R.; Li, Y.; et al. Genomic prediction in CIMMYT maize and wheat breeding programs. Heredity 2014, 112, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Combs, E.; Bernardo, R. Accuracy of Genomewide Selection for Different Traits with Constant Population Size, Heritability, and Number of Markers. Plant Genome 2013, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, R. Genome-wide selection when major genes are known. Crop Sci. 2014, 54, 68–75. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean (Range) | σ2G | σ2GxE | σ2e | H2 | LSD0.05 |
|---|---|---|---|---|---|---|
| Well-watered management | ||||||
| GY | 7.54 (4.35–11.66) | 0.54 ** | 1.19 ** | 2.02 | 0.33 | 1.12 |
| AD | 67.72 (61.5–73.5) | 2.88 ** | 4.14 ** | 2.83 | 0.51 | 3.83 |
| ASI | 0.39 (−3.0–4.5) | 0.48 ** | 0.17 ** | 1.07 | 0.58 | 1.36 |
| PH | 236.21 (155.4–292.1) | 73.02 ** | 62.05 ** | 191.62 | 0.48 | 18.02 |
| EH | 126.12 (79.9–169.9) | 63.32 ** | 67.05 ** | 82.88 | 0.54 | 17.32 |
| TLB | 2.9 (1.0–4.9) | 0.29 ** | - | 0.27382 | 0.81 | 1.03 |
| MOI | 23.41 (19.8–26.92) | 3.00 ** | 0.01 | 7.12268 | 0.63 | 5.3 |
| CR | 1.08 (0.99–3.01) | 0.02 ** | - | 0.05727 | 0.58 | 0.47 |
| GLS | 1.27 (0.91–2.92) | 0.05 ** | - | 0.15261 | 0.57 | 0.77 |
| Water stress management | ||||||
| GY | 2.7 (0.03–5.67) | 0.21 ** | - | 0.64 | 0.40 | 0.78 |
| AD | 67.31 (61.1–75.5) | 2.05 ** | - | 3.05 | 0.57 | 2.45 |
| ASI | 2.26 (−3.95–8.17) | 1.61 ** | - | 3.02 | 0.52 | 1.84 |
| PH | 176.54 (115.5–231.8) | 91.88 ** | - | 231.91 | 0.44 | 16.34 |
| EH | 91.26 (55.6–131.7) | 28.17 ** | - | 82.63 | 0.41 | 12.71 |
| MOI | 12.97 (7.8–16.5) | 1.02 * | - | 1.55 | 0.57 | 1.1 |
| SEN | 5.96 (1.91–9.14) | 0.52 ** | - | 1.33 | 0.44 | 0.9 |
| ER | 1.86 (0.02–51.1) | 12.03 ** | - | 42.71 | 0.36 | 3.78 |
| MLN management | ||||||
| MLN-DS | 6.02 (2.1–9.0) | 1.01 ** | - | 0.99 | 0.67 | 0.79 |
| AUDPC | 166.47 (75.2–264.8) | 642.8 ** | - | 453.9 | 0.74 | 20.11 |
| Trait | WW | WS | MLN |
|---|---|---|---|
| GY | 7 | 8 | - |
| AD | 16 | 12 | - |
| ASI | 7 | 8 | - |
| EH | 14 | 5 | - |
| PH | 14 | 5 | - |
| MOI | 15 | 5 | - |
| GLS | 14 | - | - |
| TLB | 11 | - | - |
| SEN | - | 11 | |
| MLNDS | - | - | 12 |
| AUDPC | - | - | 10 |
| Total | 98 | 54 | 22 |
| Trait | SNP a | Chr | p Value | MAF | Effect | Putative Candidate Gene | Predicted Function of Candidate Gene |
|---|---|---|---|---|---|---|---|
| MLN-DS | S3_184235364 | 3 | 4.74 × 10−8 | 0.21 | −0.25 | GRMZM2G429982 | Osmotin-like protein |
| S9_139517081 | 9 | 6.93 × 10−7 | 0.36 | −0.15 | GRMZM2G007514 | Protein SCAR2 | |
| S7_131111643 | 7 | 9.35 × 10−7 | 0.15 | −0.23 | GRMZM2G467907 | uncharacterized LOC100278143 | |
| S8_160531262 | 8 | 9.51 × 10−7 | 0.31 | −0.15 | GRMZM2G151614 | SUPPRESSOR OF ABI3-5 | |
| S6_139620542 | 6 | 1.52 × 10−6 | 0.15 | 0.22 | GRMZM2G061912 | uncharacterized LOC100282310 | |
| S9_3561642 | 9 | 1.79 × 10−6 | 0.18 | 0.19 | GRMZM2G110832 | uncharacterized LOC100282577 | |
| S4_166924851 | 4 | 1.94 × 10−6 | 0.29 | −0.15 | GRMZM2G141036 | Aspartyl protease family protein At5g10770 | |
| S3_144742460 | 3 | 1.98 × 10−6 | 0.28 | −0.13 | GRMZM2G062587 | Protein NETWORKED 2D | |
| S10_127441666 | 10 | 3.63 × 10−6 | 0.46 | −0.12 | GRMZM2G047370 | uncharacterized LOC100280076 | |
| S6_35895705 | 6 | 4.45 × 10−6 | 0.11 | 0.34 | GRMZM2G035502 | Glutathione dehydroascorbate reductase3 | |
| S6_38115747 | 6 | 4.50 × 10−6 | 0.09 | −0.37 | GRMZM5G818106 | phospholipase A1-II 7 | |
| S3_218903424 | 3 | 6.12 × 10−6 | 0.15 | 0.19 | GRMZM2G032244 | Adenine C2 methyltransferase pseudogene | |
| AUDPC | S1_22259426 | 1 | 3.70 × 10−9 | 0.24 | 4.81 | GRMZM2G388915 | DNA repair protein UVH3 |
| S9_139517081 | 9 | 7.48 × 10−9 | 0.36 | −5.08 | GRMZM2G007590 | Spliceosomal protein | |
| S7_131127271 | 7 | 3.26 × 10−7 | 0.15 | 5.14 | GRMZM2G467907 | uncharacterized LOC100278143 | |
| S10_125845596 | 10 | 3.56 × 10−7 | 0.28 | −4.61 | GRMZM2G003917 | fasciclin-like arabinogalactan protein 7 | |
| S4_234398586 | 4 | 3.77 × 10-7 | 0.13 | −5.64 | GRMZM2G111886 | uncharacterized LOC100383387 | |
| S6_139620542 | 6 | 4.23 × 10−7 | 0.15 | 5.86 | GRMZM2G061912 | uncharacterized LOC100282310 | |
| S8_165275778 | 8 | 7.81 × 10−7 | 0.38 | −3.59 | GRMZM2G300375 | ATP-dependent RNA helicase DHX8 | |
| S6_38115747 | 6 | 2.25 × 10−6 | 0.09 | −9.84 | GRMZM5G818106 | phospholipase A1-II 7 | |
| S2_16652265 | 2 | 5.07 × 10−6 | 0.12 | 5.50 | GRMZM2G003752 | fasciclin-like arabinogalactan protein 10 | |
| S2_3795343 | 2 | 7.82 × 10−6 | 0.27 | 6.20 | GRMZM2G321394 | protein trichome birefringence-like 8 |
| Trait | SNP a | Chr | p Value | MAF | Effect | Putative Candidate Gene | Predicted Function of Candidate Gene |
|---|---|---|---|---|---|---|---|
| GY_WW | S1_298824055 | 1 | 2.46 × 10−7 | 0.46 | −0.15 | GRMZM2G017470 | Dof zinc finger protein DOF3.6 |
| S2_213205445 | 2 | 2.17 × 10−6 | 0.24 | 0.16 | GRMZM2G030713 | O-fucosyltransferase 36 | |
| S3_13277926 | 3 | 2.62 × 10−7 | 0.41 | −0.15 | GRMZM2G026783 | uncharacterized | |
| S3_32128255 | 3 | 3.49 × 10−7 | 0.26 | −0.21 | GRMZM2G401294 | uncharacterized | |
| S4_177585108 | 4 | 8.53 × 10−7 | 0.20 | 0.20 | GRMZM2G422190 | uncharacterized | |
| S5_206615806 | 5 | 5.79 × 10−8 | 0.34 | −0.21 | GRMZM2G050734 | uncharacterized | |
| S5_208069452 | 5 | 1.03 × 10−6 | 0.39 | −0.15 | GRMZM2G421899 | uncharacterized | |
| GY_WS | S1_8635464 | 1 | 4.07 × 10−6 | 0.09 | 0.23 | GRMZM2G072814 | uncharacterized |
| S1_285928879 | 1 | 1.07 × 10−6 | 0.09 | 0.23 | GRMZM2G472167 | peptide transporter PTR2 | |
| S2_4269206 | 2 | 2.99 × 10−7 | 0.13 | 0.18 | GRMZM2G019404 | plasma-membrane H+ATPase 2 | |
| S4_168744841 | 4 | 7.33 × 10−6 | 0.13 | 0.17 | GRMZM2G422190 | uncharacterized | |
| S4_177585059 | 4 | 5.13 × 10−8 | 0.21 | 0.18 | GRMZM2G021339 | uncharacterized | |
| S6_160605809 | 6 | 1.83 × 10−6 | 0.14 | −0.19 | GRMZM2G701221 | uncharacterized | |
| S7_142907882 | 7 | 3.87 × 10−6 | 0.27 | 0.17 | GRMZM2G108133 | β-glucosidase 11 | |
| S7_157468954 | 7 | 4.61 × 10−8 | 0.21 | −0.20 | GRMZM2G134545 | dof zinc finger protein 2 |
| Trait | SNP a | Chr | p Value | MAF | Effect | Putative Candidate Gene | Predicted Function of Candidate Gene |
|---|---|---|---|---|---|---|---|
| AD_WW | S1_196052986 | 1 | 5.80 × 10−11 | 0.47 | −0.3 | GRMZM2G142383 | Uridine kinase-like protein 2 chloroplastic |
| S1_181338998 | 1 | 2.47 × 10−8 | 0.27 | −0.36 | GRMZM2G131254 | uncharacterized LOC100191530 | |
| S1_45513978 | 1 | 3.15 × 10−6 | 0.08 | 0.37 | - | uncharacterized LOC100383423 | |
| S1_77141315 | 1 | 5.83 × 10−6 | 0.14 | 0.37 | GRMZM2G425736 | uncharacterized LOC100283616 | |
| S1_85991848 | 1 | 9.18 × 10−6 | 0.44 | −0.22 | GRMZM2G040743 | putative calcium-dependent protein kinase family protein | |
| S2_164888999 | 2 | 3.56 × 10−106 | 0.06 | −0.51 | GRMZM2G151549 | eukaryotic translation initiation factor 3 subunit 6 | |
| S2_24033162 | 2 | 6.81 × 10−6 | 0.08 | −0.39 | GRMZM2G178998 | uncharacterized LOC100273446 | |
| S3_219697767 | 3 | 3.91 × 10−6 | 0.09 | −0.29 | GRMZM2G180815 | rae1-like protein | |
| S4_115254061 | 4 | 1.97 × 10−7 | 0.16 | 0.28 | GRMZM2G127690 | U-box domain-containing protein 27 | |
| S5_32847998 | 5 | 4.48 × 10−7 | 0.12 | 0.31 | GRMZM2G130580 | uncharacterized LOC100216815 | |
| S6_135702947 | 6 | 6.59 × 10−7 | 0.13 | −0.29 | GRMZM2G441565 | Mediator of RNA polymerase II transcription subunit 32 | |
| S7_15457114 | 7 | 4.55 × 10−8 | 0.18 | 0.3 | GRMZM2G028129 | uncharacterized LOC100191940 | |
| S7_172975188 | 7 | 4.84 × 10−7 | 0.12 | −0.35 | GRMZM2G158130 | uncharacterized LOC100272539 | |
| S7_174788925 | 7 | 1.37 × 10−6 | 0.47 | −0.22 | GRMZM2G006119 | corticosteroid 11-β-dehydrogenase isozyme 1 | |
| S10_88394535 | 10 | 1.09 × 10−11 | 0.3 | −0.3 | GRMZM5G848692 | uncharacterized LOC100191174 | |
| S10_87090061 | 10 | 8.84 × 10−8 | 0.06 | 0.37 | GRMZM2G028104 | 3-N-debenzoyl-2-deoxytaxol N-benzoyltransferase | |
| AD_WS | S1_70577921 | 1 | 3.41 × 10−6 | 0.28 | −0.42 | GRMZM2G049070 | E3 ubiquitin-protein ligase EL5 |
| S2_28044014 | 2 | 9.88 × 10−6 | 0.46 | 0.31 | GRMZM2G113990 | coiled-coil-helix-coiled-coil-helix domain-protein 4 | |
| S4_33885186 | 4 | 1.79 × 10−11 | 0.08 | −0.69 | GRMZM5G814904 | catechol-O-methyltransferase | |
| S5_196250543 | 5 | 6.05 × 10−6 | 0.17 | −0.33 | GRMZM2G124136 | Putative glycerol-3-phosphate transporter 4 | |
| S7_127270714 | 7 | 3.75 × 10−8 | 0.31 | −0.54 | GRMZM2G096092 | uncharacterized LOC101027214 | |
| S7_132076042 | 7 | 7.51 × 10−6 | 0.17 | 0.41 | GRMZM2G070375 | FIP1 | |
| S7_147051998 | 7 | 3.71 × 10−7 | 0.46 | −0.32 | GRMZM2G325238 | putative cysteine-rich receptor-like protein kinase 35 | |
| S7_174752364 | 7 | 2.16 × 10−7 | 0.14 | 0.44 | GRMZM2G139870 | uncharacterized LOC103633488 | |
| S8_148392640 | 8 | 1.76 × 10−11 | 0.06 | −1.01 | GRMZM2G439168 | E3 ubiquitin-protein ligase AIRP2 | |
| S9_141790219 | 9 | 2.08 × 10−6 | 0.21 | −0.34 | GRMZM2G165357 | uncharacterized LOC100193447 | |
| S9_150809900 | 9 | 9.68 × 10−7 | 0.16 | −0.45 | GRMZM2G305027 | D-type cyclin | |
| S10_85581257 | 10 | 5.47 × 10−6 | 0.49 | 0.3 | GRMZM2G060798 | uncharacterized LOC100279979 | |
| ASI_WW | S2_194040196 | 2 | 1.11 × 10−8 | 0.24 | −0.2 | GRMZM2G137541 | uncharacterized LOC100286191 |
| S2_2055642 | 2 | 2.63 × 10−6 | 0.35 | 0.1 | GRMZM2G343317 | uncharacterized LOC100274748 | |
| S2_30840909 | 2 | 9.16 × 10−6 | 0.08 | 0.3 | GRMZM2G048366 | uncharacterized LOC100194081 | |
| S3_2218652 | 3 | 3.10 × 10−6 | 0.15 | 0.16 | GRMZM2G026868 | uncharacterized LOC100276559 | |
| S5_53522965 | 5 | 8.54 × 10−8 | 0.22 | −0.2 | GRMZM2G113349 | uncharacterized LOC100191684 | |
| S6_5506422 | 6 | 7.96 × 10−6 | 0.34 | −0.12 | GRMZM2G450546 | expansin-A19 | |
| S8_3482389 | 8 | 2.45 × 10−8 | 0.3 | 0.24 | GRMZM2G136158 | Peroxidase 24 | |
| ASI_WS | S1_215203966 | 1 | 3.62 × 10−6 | 0.1 | −0.43 | GRMZM2G067235 | uncharacterized LOC100275190 |
| S1_4748614 | 1 | 7.55 × 10−7 | 0.46 | −0.26 | GRMZM2G040762 | uncharacterized LOC100381417 | |
| S2_205904889 | 2 | 2.34 x 10−6 | 0.26 | −0.28 | GRMZM2G105869 | histone-lysine N-methyltransferase SUVR3 | |
| S5_211764882 | 5 | 9.75 × 10−6 | 0.14 | −0.38 | GRMZM2G415327 | uncharacterized LOC100216930 | |
| S9_100485294 | 9 | 4.21 × 10−6 | 0.21 | −0.31 | GRMZM2G104866 | uncharacterized LOC100193380 | |
| S9_136924349 | 9 | 5.99 × 10−8 | 0.2 | 0.38 | GRMZM2G051896 | nuclear poly(A) polymerase 3 | |
| S10_135678936 | 10 | 6.72 × 10−6 | 0.44 | 0.3 | GRMZM2G016819 | Ubiquitin carboxyl-terminal hydrolase family protein | |
| S10_37346033 | 10 | 8.63 × 10−6 | 0.48 | 0.24 | GRMZM2G472703 | receptor kinase pseudogene |
| Trait | SNP a | Chr | p Value | MAF | Effect | Putative Candidate Gene | Predicted Function of Candidate Gene |
|---|---|---|---|---|---|---|---|
| PH_WW | S1_234263371 | 1 | 2.52 × 10−6 | 0.39 | 1.63 | GRMZM2G153233 | uncharacterized LOC100304210 |
| S1_32087637 | 1 | 1.57 × 10−8 | 0.5 | −2.04 | GRMZM2G100629 | uncharacterized LOC100277213 | |
| S1_51399301 | 1 | 4.64 × 10−8 | 0.27 | −2.60 | GRMZM5G851485 | uncharacterized LOC100274900 | |
| S2_12352637 | 2 | 2.32 × 10−7 | 0.26 | 2.29 | GRMZM2G156356 | maltose excess protein 1-like | |
| S3_13264837 | 3 | 5.95 × 10−7 | 0.24 | 2.00 | GRMZM2G026783 | uncharacterized LOC100278056 | |
| S3_166807659 | 3 | 1.52 × 10−8 | 0.39 | 2.30 | GRMZM2G366142 | uncharacterized LOC100193554 | |
| S4_228623517 | 4 | 2.49 × 10−6 | 0.35 | −1.73 | GRMZM2G016923 | uncharacterized LOC100502389 | |
| S5_27226539 | 5 | 7.78 × 10−12 | 0.24 | 3.31 | GRMZM2G428356 | uncharacterized LOC100277327 | |
| S6_161804186 | 6 | 2.08 × 10−9 | 0.25 | −2.64 | GRMZM2G170625 | Jacalin-related lectin 3 | |
| S7_163790932 | 7 | 2.10 × 10−7 | 0.08 | −3.81 | GRMZM2G106548 | scarecrow-like protein 23 | |
| S7_163967764 | 7 | 2.75 × 10−9 | 0.36 | −2.32 | GRMZM2G006942 | exocyst complex component EXO84C | |
| S7_164656257 | 7 | 7.38 × 10−6 | 0.23 | −1.78 | GRMZM2G037545 | uncharacterized LOC100217043 | |
| S8_14844357 | 8 | 3.24 × 10−10 | 0.15 | 3.67 | GRMZM2G052869 | metallothionein-like protein 2A | |
| S8_67856497 | 8 | 3.49 × 10−6 | 0.09 | −3.04 | GRMZM2G416216 | uncharacterized LOC100384078 | |
| PH_WS | S2_18001352 | 2 | 6.22 × 10−7 | 0.21 | 3.52 | GRMZM2G318956 | uncharacterized LOC100381487 |
| S2_43203188 | 2 | 6.87 × 10−6 | 0.15 | 3.09 | GRMZM2G114523 | lysine histidine transporter-like 6 | |
| S7_160313368 | 7 | 3.28 × 10−7 | 0.36 | −2.85 | GRMZM2G431039 | glucan endo-1,3-β-glucosidase 13 | |
| S7_162003719 | 7 | 7.44 × 10−6 | 0.25 | −3.11 | GRMZM2G153162 | eukaryotic translation initiation factor 4G | |
| S8_158986117 | 8 | 4.46 × 10−9 | 0.23 | 4.22 | GRMZM2G057416 | uncharacterized LOC100216812 | |
| EH_WW | S1_5744898 | 1 | 2.81 × 10−7 | 0.13 | 2.23 | GRMZM2G025642 | uncharacterized LOC100383060 |
| S2_184012021 | 2 | 5.92 × 10−6 | 0.22 | 1.4 | GRMZM2G116196 | AUGMIN subunit 5 | |
| S2_54204575 | 2 | 4.86 × 10−7 | 0.22 | −1.84 | GRMZM2G135727 | 60S ribosomal protein L3 | |
| S2_6821849 | 2 | 6.13 × 10−7 | 0.48 | −1.37 | GRMZM2G063519 | putative galacturonosyltransferase 10 | |
| S3_103574552 | 3 | 1.20 × 10−6 | 0.11 | 1.75 | GRMZM2G147811 | serine/threonine-protein kinase prpf4B-like | |
| S3_15433905 | 3 | 8.42 × 10−11 | 0.18 | −2.5 | GRMZM2G162182 | uncharacterized LOC103651780 | |
| S4_141091471 | 4 | 8.91 × 10−7 | 0.28 | −1.56 | GRMZM2G179810 | Adenine phosphoribosyltransferase 2 | |
| S4_166924899 | 4 | 3.39 × 10−11 | 0.37 | 2.48 | GRMZM2G141036 | aspartyl protease family protein At5g10770 | |
| S4_230932334 | 4 | 2.10 × 10−6 | 0.26 | −1.77 | GRMZM2G004835 | uncharacterized LOC100273059 | |
| S5_182542047 | 5 | 1.39 × 10−7 | 0.49 | 1.69 | GRMZM2G121236 | umecyanin | |
| S5_205332507 | 5 | 1.33 × 10−8 | 0.16 | 2.47 | - | uncharacterized LOC100502221 | |
| S5_22544395 | 5 | 3.48 × 10−6 | 0.49 | 1.29 | GRMZM2G059013 | fringe-related protein | |
| S6_96126766 | 6 | 1.51 × 10−6 | 0.29 | −1.41 | GRMZM2G088086 | uncharacterized LOC100286007 | |
| S10_138873729 | 10 | 1.43 × 10−7 | 0.25 | −1.64 | GRMZM2G155776 | uncharacterized LOC100276813 | |
| EH_WS | S2_195919610 | 2 | 2.45 × 10−7 | 0.44 | −2.06 | GRMZM2G365374 | heat shock 70 kDa protein |
| S2_5904286 | 2 | 2.19 × 10−7 | 0.37 | −1.84 | GRMZM2G372102 | 36.4 kDa proline-rich protein | |
| S7_87194068 | 7 | 2.01 × 10−7 | 0.4 | −1.69 | GRMZM2G157953 | NAD(P)H dehydrogenase subunit CRR3 chloroplast | |
| S9_118825634 | 9 | 2.55 × 10−7 | 0.17 | −2.24 | GRMZM2G108619 | uncharacterized LOC100275618 | |
| S9_150868777 | 9 | 6.71 × 10−6 | 0.39 | −1.31 | GRMZM2G006721 | D-type cyclin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadessa, K.; Beyene, Y.; Ifie, B.E.; Suresh, L.M.; Olsen, M.S.; Ogugo, V.; Wegary, D.; Tongoona, P.; Danquah, E.; Offei, S.K.; et al. Identification of Genomic Regions Associated with Agronomic and Disease Resistance Traits in a Large Set of Multiple DH Populations. Genes 2022, 13, 351. https://doi.org/10.3390/genes13020351
Sadessa K, Beyene Y, Ifie BE, Suresh LM, Olsen MS, Ogugo V, Wegary D, Tongoona P, Danquah E, Offei SK, et al. Identification of Genomic Regions Associated with Agronomic and Disease Resistance Traits in a Large Set of Multiple DH Populations. Genes. 2022; 13(2):351. https://doi.org/10.3390/genes13020351
Chicago/Turabian StyleSadessa, Kassahun, Yoseph Beyene, Beatrice E. Ifie, L. M. Suresh, Michael S. Olsen, Veronica Ogugo, Dagne Wegary, Pangirayi Tongoona, Eric Danquah, Samuel Kwame Offei, and et al. 2022. "Identification of Genomic Regions Associated with Agronomic and Disease Resistance Traits in a Large Set of Multiple DH Populations" Genes 13, no. 2: 351. https://doi.org/10.3390/genes13020351
APA StyleSadessa, K., Beyene, Y., Ifie, B. E., Suresh, L. M., Olsen, M. S., Ogugo, V., Wegary, D., Tongoona, P., Danquah, E., Offei, S. K., Prasanna, B. M., & Gowda, M. (2022). Identification of Genomic Regions Associated with Agronomic and Disease Resistance Traits in a Large Set of Multiple DH Populations. Genes, 13(2), 351. https://doi.org/10.3390/genes13020351