Gene Ontology Groups and Signaling Pathways Regulating the Process of Avian Satellite Cell Differentiation


 ,
,  , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Material and Methods
2.1. Animals
2.2. The Study Design
2.2.1. Isolation, Culture and Differentiation of Satellite Cells
2.2.2. RNA Isolation
2.2.3. RNAseq
2.2.4. Bioinformatics Analysis
2.2.5. Overall Enrichment Analysis
2.2.6. Protein–Protein Interaction (PPI) Networks
2.2.7. Assignment of Differentially Expressed Genes
2.2.8. Signalling Pathways Analysis
2.2.9. Ingenuity Pathway Analysis
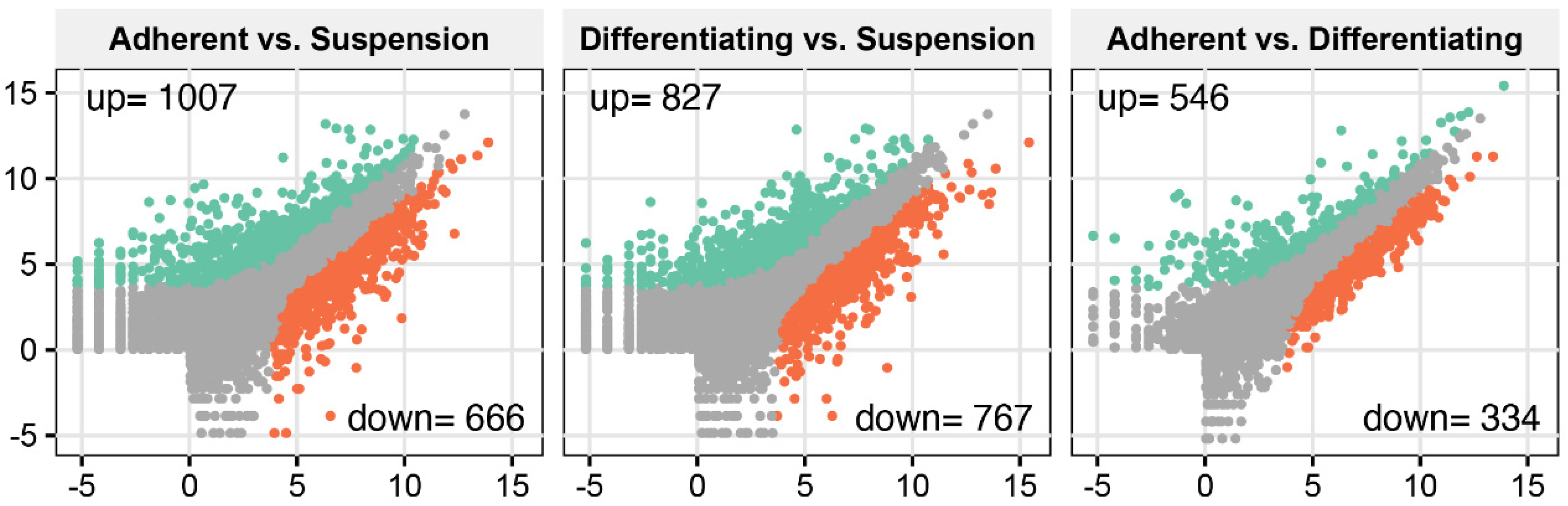
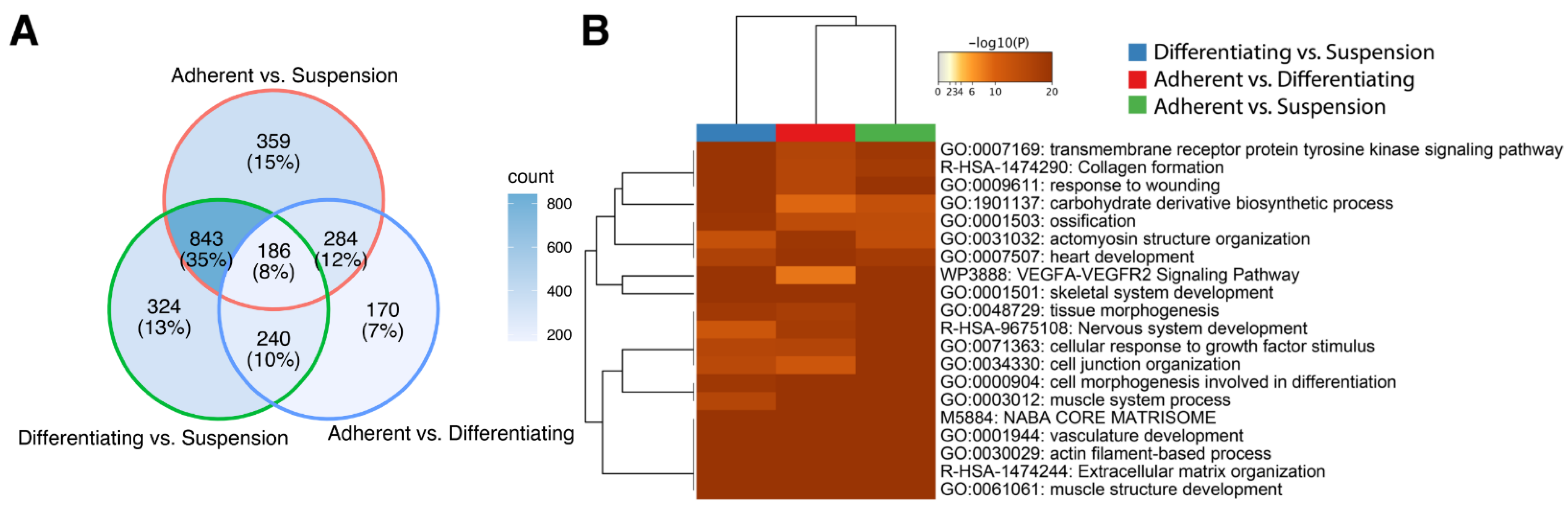
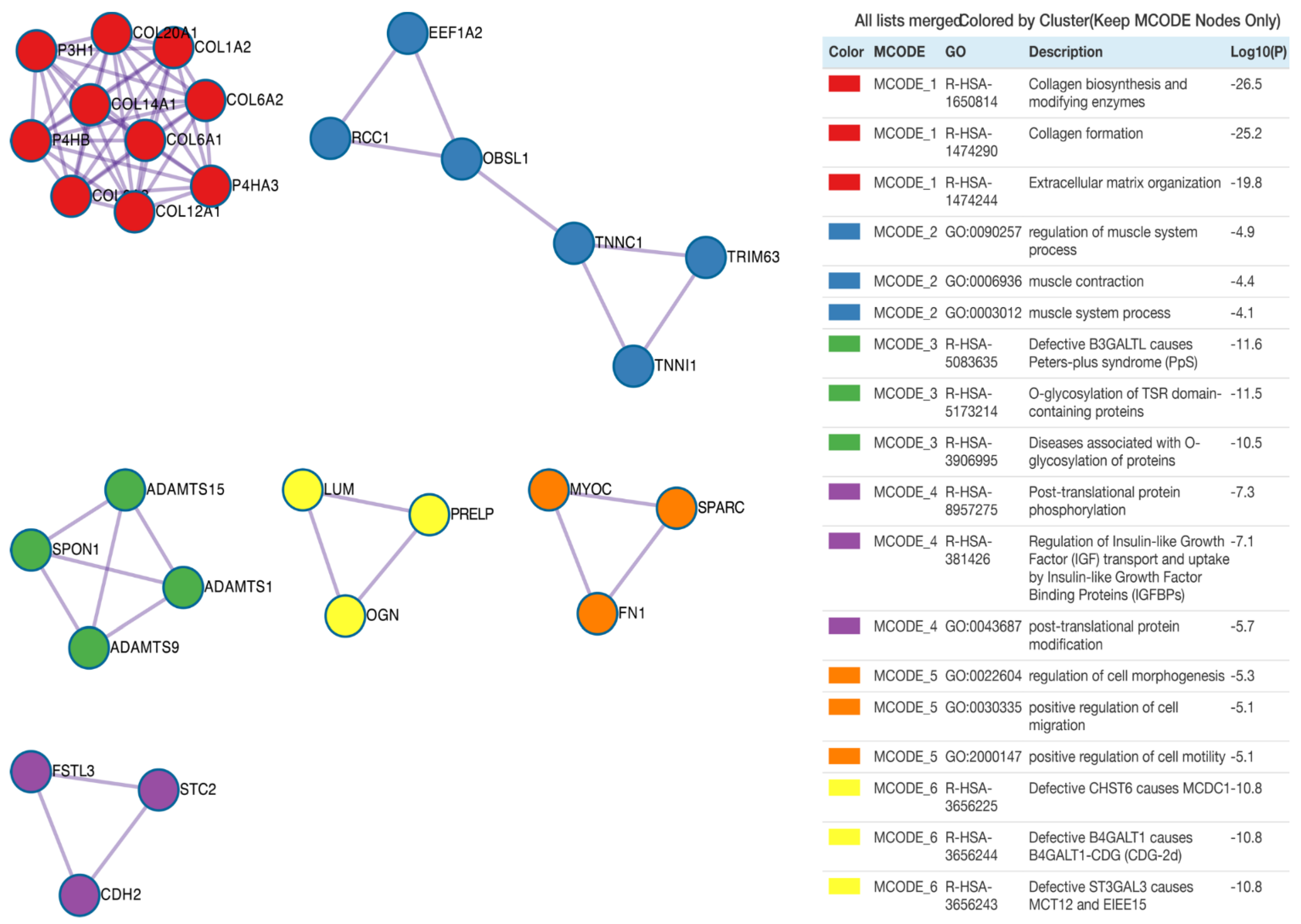
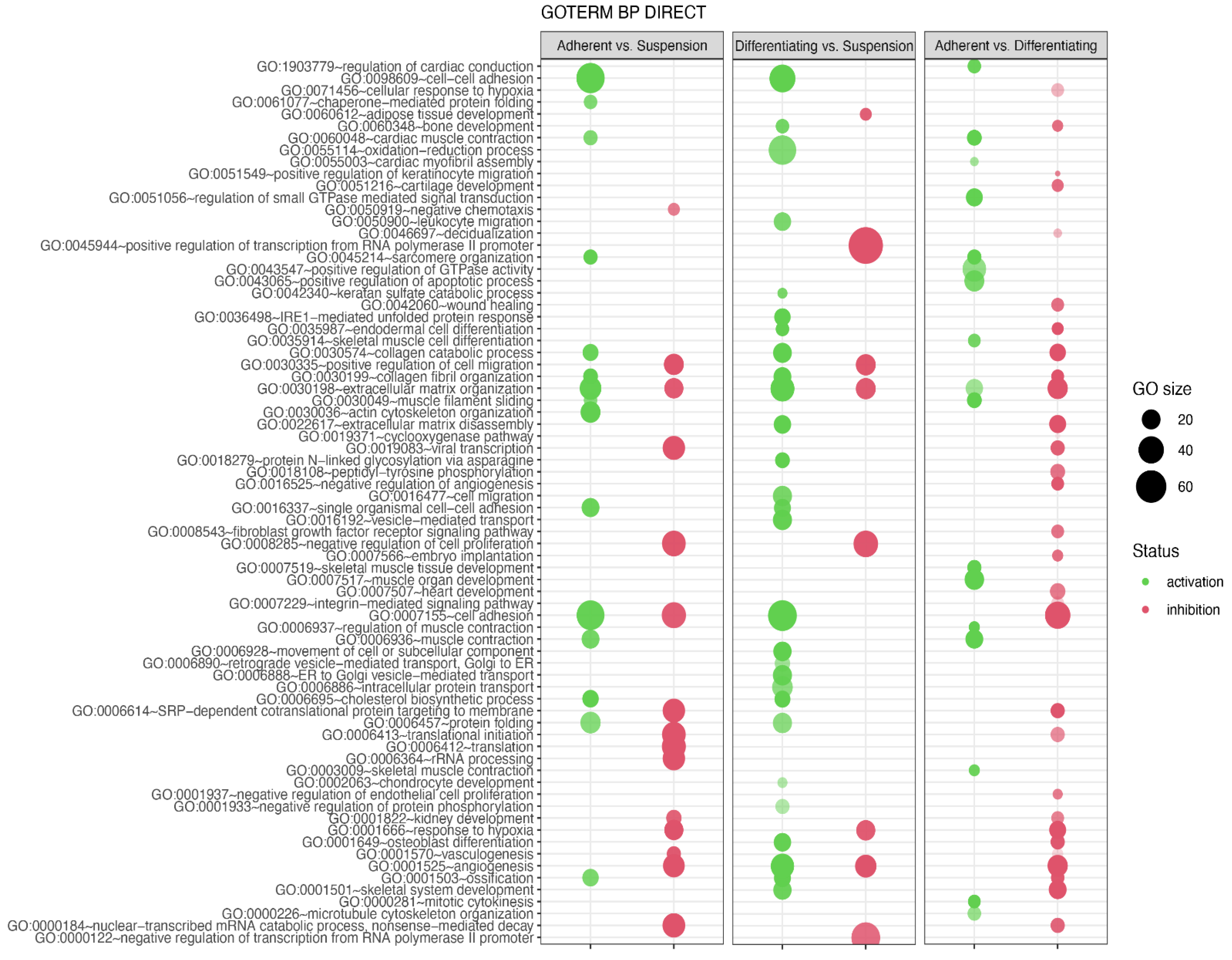
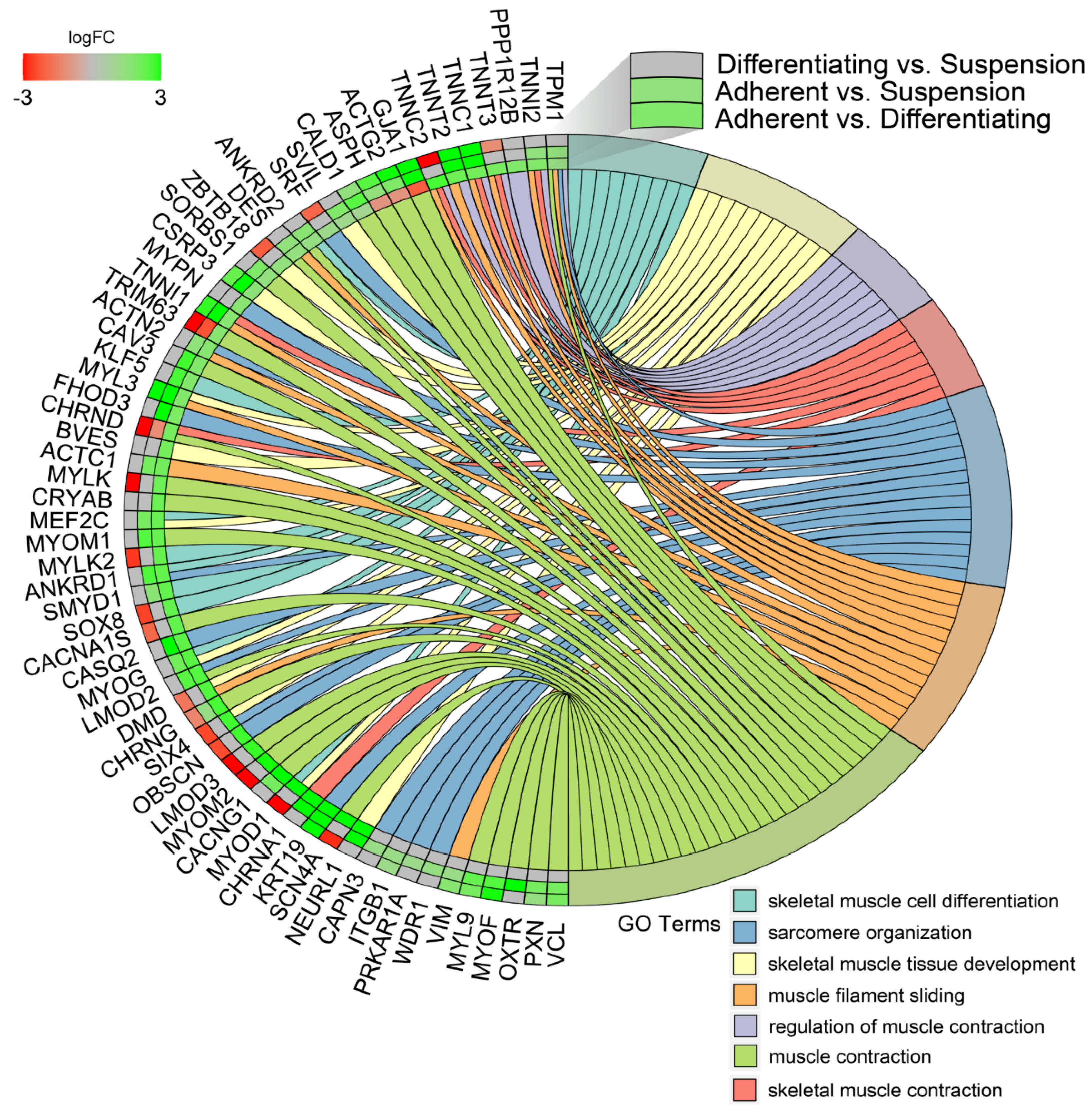
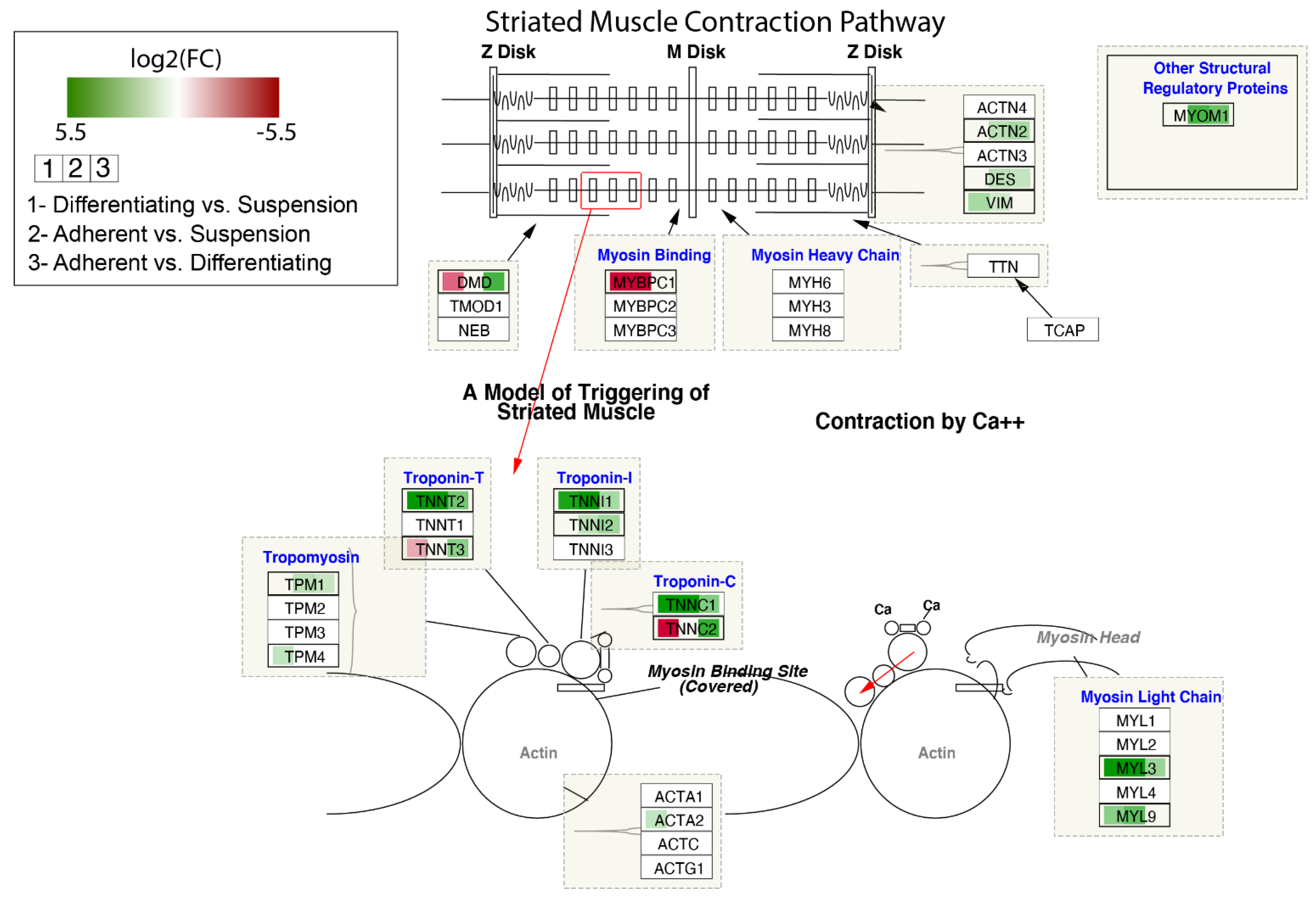
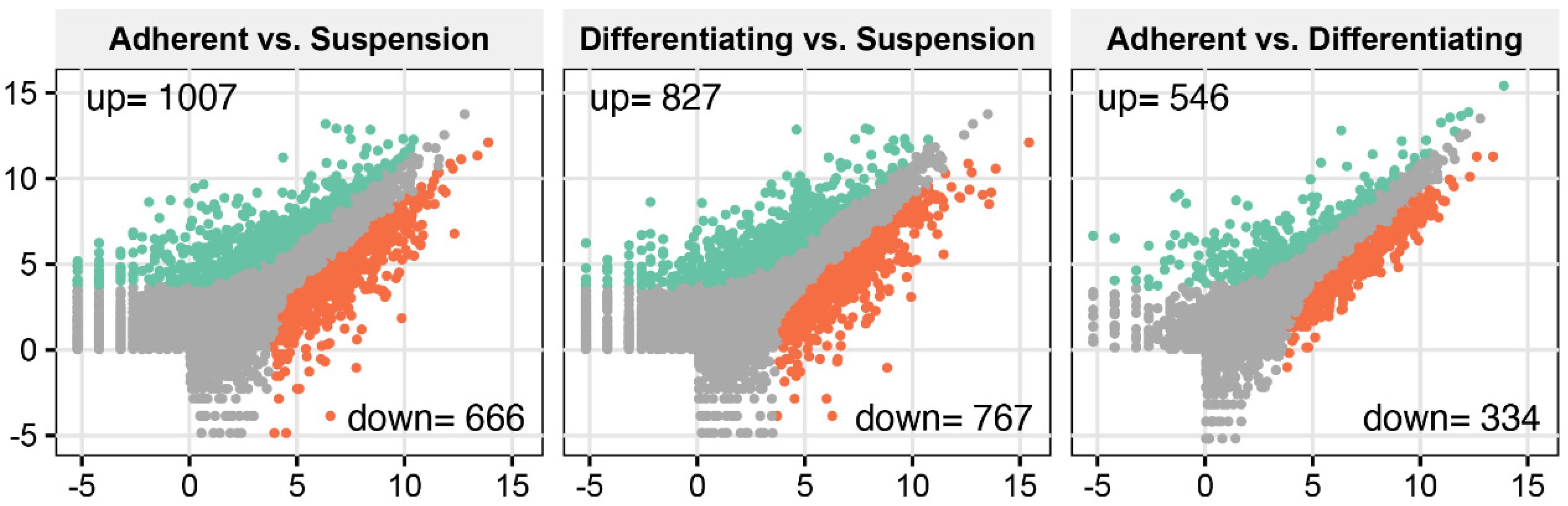
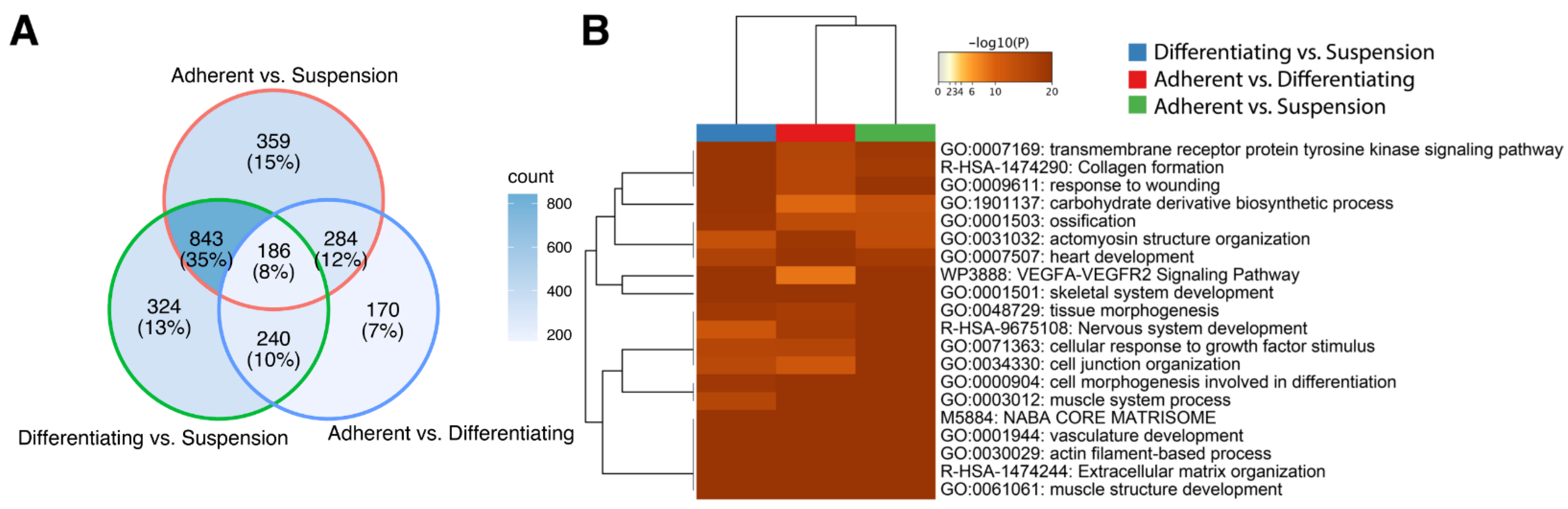
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mekonnen, M.M.; Hoekstra, A.Y. A Global Assessment of the Water Footprint of Farm Animal Products. Ecosystems 2012, 15, 401–415. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, A.Y. Water for animal products: A blind spot in water policy. Environ. Res. Lett. 2014, 9, 091003. [Google Scholar] [CrossRef] [Green Version]
- Rzymski, P.; Kulus, M.; Jankowski, M.; Dompe, C.; Bryl, R.; Petitte, J.N.; Kempisty, B.; Mozdziak, P. COVID-19 pandemic is a call to search for alternative protein sources as food and feed: A review of possibilities. Nutrients 2021, 13, 150. [Google Scholar] [CrossRef] [PubMed]
- Hocquette, J.F. Is in vitro meat the solution for the future? Meat Sci. 2016, 120, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Wilks, M.; Phillips, C.J.C. Attitudes to in vitro meat: A survey of potential consumers in the United States. PLoS ONE 2017, 12, e0171904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, Z.F.; Kumar, S.; Fayaz, H. In vitro meat production: Challenges and benefits over conventional meat production. J. Integr. Agric. 2015, 14, 241–248. [Google Scholar] [CrossRef]
- Sharma, S.; Thind, S.S.; Kaur, A. In vitro meat production system: Why and how? J. Food Sci. Technol. 2015, 52, 7599–7607. [Google Scholar] [CrossRef] [Green Version]
- Stephens, N.; Dunsford, I.; di Silvio, L.; Ellis, M.; Glencross, A.; Sexton, A.E. Bringing cultured meat to market: Technical, socio-political, and regulatory challenges in cellular agriculture. Trends Food Sci. Technol. 2018, 78, 155–166. [Google Scholar] [CrossRef]
- Datar, I.; Betti, M. Possibilities for an in vitro meat production system. Innov. Food Sci. Emerg. Technol. 2010, 11, 13–22. [Google Scholar] [CrossRef]
- Woll, S. On visions and promises—Ethical aspects of in vitro meat. Emerg. Top. Life Sci. 2019, 3, 753–758. [Google Scholar] [CrossRef]
- Narenji, A.G.; Petitte, J.N.; Kulus, M.; Stefańska, K.; Perek, J.; Kulus, J.; Wieczorkiewicz, M.; Jankowski, M.; Bryja, A.; Bryl, R.; et al. Telomerase Activity and Myogenesis Ability as an Indicator of Cultured Turkey Satellite Cell Ability for in Vitro Meat Production. Med. J. Cell Biol. 2021, 9, 19–26. [Google Scholar] [CrossRef]
- Jankowski, M.; Mozdziak, P.; Petitte, J.; Kulus, M.; Kempisty, B. Avian satellite cell plasticity. Animals 2020, 10, 1322. [Google Scholar] [CrossRef] [PubMed]
- Leggett, R.M.; Ramirez-Gonzalez, R.H.; Clavijo, B.J.; Waite, D.; Davey, R.P. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Front. Genet. 2013, 4, 288. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Di Pietro, A.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Wernersson, R.; Hansen, R.B.; Horn, H.; Mercer, J.; Slodkowicz, G.; Workman, C.T.; Rigina, O.; Rapacki, K.; Stærfeldt, H.H.; et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat. Methods 2016, 14, 61–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Fresno, C.; Fernández, E.A. RDAVIDWebService: A versatile R interface to DAVID. Bioinformatics 2013, 29, 2810–2811. [Google Scholar] [CrossRef] [Green Version]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; DIgles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Gustavsen, J.A.; Pai, S.; Isserlin, R.; Demchak, B.; Pico, A.R. RCy3: Network biology using Cytoscape from within R. F1000Research 2019, 8, 1774. [Google Scholar] [CrossRef]
- Cohen, J. A Coefficient of Agreement for Nominal Scales. Educ. Psychol. Meas. 1960, 20, 37–46. [Google Scholar] [CrossRef]
- Oñate, B.; Vilahur, G.; Camino-López, S.; Díez-Caballero, A.; Ballesta-López, C.; Ybarra, J.; Moscatiello, F.; Herrero, J.; Badimon, L. Stem cells isolated from adipose tissue of obese patients show changes in their transcriptomic profile that indicate loss in stemcellness and increased commitment to an adipocyte-like phenotype. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budna-Tukan, J.; Światły-Błaszkiewicz, A.; Celichowski, P.; Kałużna, S.; Konwerska, A.; Sujka-Kordowska, P.; Jankowski, M.; Kulus, M.; Jeseta, M.; Piotrowska-Kempisty, H.; et al. “Biological Adhesion” is a Significantly Regulated Molecular Process during Long-Term Primary In Vitro Culture of Oviductal Epithelial Cells (Oecs): A Transcriptomic and Proteomic Study. Int. J. Mol. Sci. 2019, 20, 3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dompe, C.; Kranc, W.; Jopek, K.; Kowalska, K.; Ciesiółka, S.; Chermuła, B.; Bryja, A.; Jankowski, M.; Perek, J.; Józkowiak, M.; et al. Muscle Cell Morphogenesis, Structure, Development and Differentiation Processes Are Significantly Regulated during Human Ovarian Granulosa Cells In Vitro Cultivation. J. Clin. Med. 2020, 9, 2006. [Google Scholar] [CrossRef] [PubMed]
- Adomavicius, T.; Guaita, M.; Zhou, Y.; Jennings, M.D.; Latif, Z.; Roseman, A.M.; Pavitt, G.D. The structural basis of translational control by eIF2 phosphorylation. Nat. Commun. 2019, 10, 2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeske, R.; Yuan, X.; Fu, Q.; Bunnell, B.A.; Logan, T.M.; Li, Y. In Vitro Culture Expansion Shifts the Immune Phenotype of Human Adipose-Derived Mesenchymal Stem Cells. Front. Immunol. 2021, 12, 621744. [Google Scholar] [CrossRef] [PubMed]
- Bijonowski, B.M.; Fu, Q.; Yuan, X.; Irianto, J.; Li, Y.; Grant, S.C.; Ma, T. Aggregation-induced integrated stress response rejuvenates culture-expanded human mesenchymal stem cells. Biotechnol. Bioeng. 2020, 117, 3136–3149. [Google Scholar] [CrossRef]
- Kanojia, D.; Zhou, W.; Zhang, J.; Jie, C.; Lo, P.K.; Wang, Q.; Chen, H. Proteomic profiling of cancer stem cells derived from primary tumors of HER2/Neu transgenic mice. Proteomics 2012, 12, 3407–3415. [Google Scholar] [CrossRef]
- Proud, C.G. mTOR signalling in health and disease. In Biochemical Society Transactions; Portland Press: London, UK, 2011; Volume 39, pp. 431–436. [Google Scholar]
- Bodine, S.C. mTOR Signaling and the Molecular Adaptation to Resistance Exercise. Med. Sci. Sport Exerc. 2006, 38, 1950–1957. [Google Scholar] [CrossRef]
- Gharibi, B.; Ghuman, M.; Hughes, F.J. DDIT4 regulates mesenchymal stem cell fate by mediating between HIF1α and mTOR signalling. Sci. Rep. 2016, 6, 36889. [Google Scholar] [CrossRef] [Green Version]
- Allan, S. Seeing mTOR in a new light. Nat. Rev. Immunol. 2008, 8, 904. [Google Scholar] [CrossRef]
- Shrestha, N.; Bahnan, W.; Wiley, D.J.; Barber, G.; Fields, K.A.; Schesser, K. Eukaryotic initiation factor 2 (eIF2) signaling regulates proinflammatory cytokine expression and bacterial invasion. J. Biol. Chem. 2012, 287, 28738–28744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, O.H.; Michalak, M.; Verkhratsky, A. Calcium signalling: Past, present and future. Cell Calcium 2005, 38, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Baylor, S.M.; Hollingworth, S. Calcium indicators and calcium signalling in skeletal muscle fibres during excitation-contraction coupling. Prog. Biophys. Mol. Biol. 2011, 105, 162–179. [Google Scholar] [CrossRef] [Green Version]
- Tu, M.K.; Levin, J.B.; Hamilton, A.M.; Borodinsky, L.N. Calcium signaling in skeletal muscle development, maintenance and regeneration. Cell Calcium 2016, 59, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumi, R.; Wuollet, A.L.; Tabata, K.; Nishimura, S.; Tabata, S.; Mizunoya, W.; Ikeuchi, Y.; Allen, R.E. A role for calcium-calmodulin in regulating nitric oxide production during skeletal muscle satellite cell activation. Am. J. Physiol.-Cell Physiol. 2009, 296, 922–929. [Google Scholar] [CrossRef]
- Tonelli, F.M.P.; Santos, A.K.; Gomes, K.N.; Ladeira, L.O.; Resende, R.R.; Gomes, D.A.; Da Silva, S.L. Stem cells and calcium signaling. Adv. Exp. Med. Biol. 2012, 740, 891–916. [Google Scholar] [CrossRef] [Green Version]
- Xiaoping, C.; Yong, L. Role of matrix metalloproteinases in skeletal muscle: Migration, differentiation, regeneration and fibrosis. Cell Adhes. Migr. 2009, 3, 337–341. [Google Scholar]
- Yamada, M.; Tatsumi, R.; Kikuiri, T.; Okamoto, S.; Nonoshita, S.; Mizunoya, W.; Ikeuchi, Y.; Shimokawa, H.; Sunagawa, K.; Allen, R.E. Matrix metalloproteinases are involved in mechanical stretch-induced activation of skeletal muscle satellite cells. Muscle Nerve 2006, 34, 313–319. [Google Scholar] [CrossRef]
- Nishimura, T.; Kazuki, A.E.; Ae, N.; Ae, Y.K.; Kato-Mori, Y.; Jun-Ichi, A.E.; Ae, W.; Hattori, A. Inhibition of matrix metalloproteinases suppresses the migration of skeletal muscle cells. J. Muscle Res. Cell Motil. 2008, 29, 37–44. [Google Scholar] [CrossRef]
- Mannello, F.; Tonti, G.A.M.; Bagnara, G.P.; Papa, S. Role and Function of Matrix Metalloproteinases in the Differentiation and Biological Characterization of Mesenchymal Stem Cells. Stem Cells 2006, 24, 475–481. [Google Scholar] [CrossRef]
- Almalki, S.G.; Agrawal, D.K. Effects of matrix metalloproteinases on the fate of mesenchymal stem cells. Stem Cell Res. Ther. 2016, 7, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessenbrock, K.; Wang, C.Y.; Werb, Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 2015, 44–46, 184–190. [Google Scholar] [CrossRef]
- Bae, J.H.; Hong, M.; Jeong, H.J.; Kim, H.; Lee, S.J.; Ryu, D.; Bae, G.U.; Cho, S.C.; Lee, Y.S.; Krauss, R.S.; et al. Satellite cell-specific ablation of Cdon impairs integrin activation, FGF signalling, and muscle regeneration. J. Cachexia Sarcopenia Muscle 2020, 11, 1089–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prowse, A.B.J.; Chong, F.; Gray, P.P.; Munro, T.P. Stem cell integrins: Implications for ex-vivo culture and cellular therapies. Stem Cell Res. 2011, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brizzi, M.F.; Tarone, G.; Defilippi, P. Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr. Opin. Cell Biol. 2012, 24, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.D. The Dynamic Actin Cytoskeleton in Smooth Muscle. In Advances in Pharmacology; Academic Press Inc.: Cambridge, MA, USA, 2018; Volume 81, pp. 1–38. [Google Scholar]
- Kee, A.J.; Gunning, P.W.; Hardeman, E.C. Diverse roles of the actin cytoskeleton in striated muscle. J. Muscle Res. Cell Motil. 2009, 30, 187–197. [Google Scholar] [CrossRef]
- Tang, D.D.; Gerlach, B.D. The roles and regulation of the actin cytoskeleton, intermediate filaments and microtubules in smooth muscle cell migration. Respir. Res. 2017, 18, 54. [Google Scholar] [CrossRef] [Green Version]
- Slot, I.G.M.; Schols, A.M.W.J.; Vosse, B.A.H.; Kelders, M.C.J.M.; Gosker, H.R. Hypoxia differentially regulates muscle oxidative fiber type and metabolism in a HIF-1α-dependent manner. Cell Signal. 2014, 26, 1837–1845. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Lee, D.S.M.; Skuli, N.; Mesquita, R.C.; Kim, M.N.; Yodh, A.G.; Nguyen-McCarty, M.; Li, B.; Simon, M.C. HIF modulation of wnt signaling regulates skeletal myogenesis in vivo. Development 2015, 142, 2405–2412. [Google Scholar] [CrossRef] [Green Version]
- Valle-Tenney, R.; Rebolledo, D.; Acuña, M.J.; Brandan, E. HIF-hypoxia signaling in skeletal muscle physiology and fibrosis. J. Cell Commun. Signal. 2020, 14, 147–158. [Google Scholar] [CrossRef]
- Li, X.; Zhu, L.L.; Chen, X.P.; Fan, M. Role of HIF signaling pathway involved in effects of hypoxia on proliferation and differentiation of myoblasts. Sheng Li Ke Xue Jin Zhan 2007, 38, 224–228. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adherent vs. Suspension | ||||||
|---|---|---|---|---|---|---|
| Symbol | Description | RPKM (Adherent) | RPKM (Suspension) | M | D | p-Value |
| CACNG3 | calcium voltage-gated channel auxiliary subunit γ 3 | 93.74 | 0.07 | 10.40 | 93.67 | 0.0004 |
| WNT10A | Wnt family member 10A | 22.51 | 0.03 | 9.34 | 22.48 | 0.01 |
| ADAM8 | ADAM metallopeptidase domain 8 | 215.70 | 0.49 | 8.79 | 215.21 | 0.00003 |
| CHST1 | carbohydrate sulfotransferase 1 | 15.41 | 0.03 | 8.79 | 15.37 | 0.03 |
| MYL3 | myosin light chain 3 | 932.39 | 3.61 | 8.01 | 928.79 | 0.0000001 |
| GJD4 | gap junction protein delta 4 | 34.45 | 0.21 | 7.37 | 34.24 | 0.005 |
| NMU | neuromedin U | 32.12 | 0.21 | 7.27 | 31.91 | 0.006 |
| OBSL1 | obscurin like cytoskeletal adaptor 1 | 219.66 | 1.53 | 7.17 | 218.13 | 0.00003 |
| WNT7A | Wnt family member 7A | 17.79 | 0.14 | 7.00 | 17.66 | 0.02 |
| MMP27 | matrix metallopeptidase 27 | 79.04 | 0.62 | 6.98 | 78.42 | 0.0007 |
| IFITM5 | interferon induced transmembrane protein 5 | 0.16 | 117.07 | −9.49 | 116.90 | 0.0002 |
| SPON2 | spondin 2 | 0.54 | 426.83 | −9.62 | 426.29 | 0.0000001 |
| CDH19 | cadherin 19 | 0.03 | 24.56 | −9.82 | 24.54 | 0.01 |
| DPT | dermatopontin | 0.05 | 51.28 | −9.88 | 51.23 | 0.002 |
| SNCA | synuclein α | 0.03 | 29.08 | −10.07 | 29.05 | 0.008 |
| CHODL | chondrolectin | 0.03 | 31.02 | −10.16 | 30.99 | 0.007 |
| SCARA3 | scavenger receptor class A member 3 | 0.03 | 36.43 | −10.39 | 36.40 | 0.004 |
| PLP1 | proteolipid protein 1 | 0.05 | 75.57 | −10.44 | 75.51 | 0.0007 |
| C1QTNF2 | C1q and TNF related 2 | 0.05 | 75.98 | −10.45 | 75.93 | 0.0007 |
| FGL2 | fibrinogen like 2 | 0.27 | 397.62 | −10.52 | 397.35 | 0.0000001 |
| Differentiating vs. Suspension | ||||||
| CACNG3 | calcium voltage-gated channel auxiliary subunit γ 3 | 77.22 | 0.07 | 10.12 | 77.15 | 0.0008 |
| ADAM8 | ADAM metallopeptidase domain 8 | 455.09 | 0.49 | 9.87 | 454.61 | 0.0000001 |
| MMP13 | matrix metallopeptidase 13 | 64.49 | 0.14 | 8.86 | 64.35 | 0.001 |
| EPHX4 | epoxide hydrolase 4 | 13.12 | 0.07 | 7.56 | 13.05 | 0.04 |
| RSPO4 | R-spondin 4 | 23.03 | 0.14 | 7.37 | 22.89 | 0.01 |
| MMP27 | matrix metallopeptidase 27 | 90.79 | 0.62 | 7.18 | 90.16 | 0.0005 |
| ACAN | agg recan | 92.72 | 0.69 | 7.06 | 92.03 | 0.0005 |
| PHGDH | phosphoglycerate dehydrogenase | 980.38 | 8.54 | 6.84 | 971.84 | 0.0000001 |
| PRRX2 | paired related homeobox 2 | 101.80 | 1.18 | 6.43 | 100.62 | 0.0004 |
| PGF | placental growth factor | 227.74 | 2.91 | 6.29 | 224.83 | 0.00003 |
| MMRN2 | multimerin 2 | 0.06 | 28.66 | −9.02 | 28.60 | 0.008 |
| SNCA | synuclein α | 0.06 | 29.08 | −9.04 | 29.02 | 0.008 |
| MATN1 | matrilin 1 | 0.03 | 17.21 | −9.28 | 17.18 | 0.02 |
| RGS5 | regulator of G-protein signaling 5 | 0.11 | 68.98 | −9.28 | 68.87 | 0.001 |
| BCAS1 | breast carcinoma amplified sequence 1 | 0.17 | 104.16 | −9.29 | 103.99 | 0.0004 |
| GJA4 | gap junction protein α 4 | 0.03 | 17.63 | −9.31 | 17.60 | 0.02 |
| CNTN1 | contactin 1 | 0.06 | 38.58 | −9.44 | 38.53 | 0.004 |
| FOXJ1 | forkhead box J1 | 0.03 | 27.48 | −9.96 | 27.45 | 0.009 |
| FGL2 | fibrinogen like 2 | 0.22 | 397.63 | −10.81 | 397.41 | 0.0000001 |
| PLP1 | proteolipid protein 1 | 0.03 | 75.57 | −11.41 | 75.54 | 0.0009 |
| Adherent vs. Differentiating | ||||||
| OPCML | opioid binding protein/cell adhesion molecule like | 14.11 | 0.50 | 4.82 | 13.61 | 0.04 |
| FAT3 | FAT atypical cadherin 3 | 27.89 | 1.11 | 4.65 | 26.78 | 0.008 |
| PDGFB | platelet derived growth factor subunit B | 26.21 | 1.11 | 4.56 | 25.10 | 0.01 |
| OPTC | opticin | 34.67 | 1.66 | 4.38 | 33.01 | 0.005 |
| SLIT1 | slit guidance ligand 1 | 504.98 | 28.24 | 4.16 | 476.74 | 0.00006 |
| PLXNA2 | plexin A2 | 15.03 | 0.89 | 4.08 | 14.15 | 0.03 |
| PAX7 | paired box 7 | 84.54 | 5.26 | 4.01 | 79.28 | 0.0007 |
| PTPRQ | protein tyrosine phosphatase receptor type Q | 80.53 | 5.04 | 4.00 | 75.49 | 0.0008 |
| NHSL2 | NHS like 2 | 53.83 | 3.88 | 3.80 | 49.95 | 0.002 |
| AIF1L | allograft inflammatory factor 1 like | 84.71 | 6.59 | 3.68 | 78.12 | 0.0008 |
| SCIN | scinderin | 0.11 | 24.97 | −7.85 | 24.86 | 0.01 |
| GFPT2 | glutamine-fructose-6-phosphate transaminase 2 | 0.38 | 105.42 | −8.12 | 105.04 | 0.0002 |
| SLC1A6 | solute carrier family 1 member 6 | 0.05 | 16.67 | −8.26 | 16.61 | 0.02 |
| TTC29 | tetratricopeptide repeat domain 29 | 0.16 | 67.94 | −8.71 | 67.77 | 0.0009 |
| SPON2 | spondin 2 | 0.54 | 377.28 | −9.44 | 376.74 | 0.0000001 |
| OGN | osteoglycin | 0.43 | 546.16 | −10.30 | 545.72 | 0.0000001 |
| AKR1D1 | aldo-keto reductase family 1 member D1 | 0.38 | 480.65 | −10.31 | 480.27 | 0.0000001 |
| DPT | dermatopontin | 0.05 | 90.36 | −10.70 | 90.31 | 0.0003 |
| ACAN | aggrecan | 0.05 | 92.74 | −10.74 | 92.69 | 0.0003 |
| SCARA3 | scavenger receptor class A member 3 | 0.03 | 101.21 | −11.87 | 101.19 | 0.0002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golkar-Narenji, A.; Antosik, P.; Nolin, S.; Rucinski, M.; Jopek, K.; Zok, A.; Sobolewski, J.; Jankowski, M.; Zdun, M.; Bukowska, D.; et al. Gene Ontology Groups and Signaling Pathways Regulating the Process of Avian Satellite Cell Differentiation. Genes 2022, 13, 242. https://doi.org/10.3390/genes13020242
Golkar-Narenji A, Antosik P, Nolin S, Rucinski M, Jopek K, Zok A, Sobolewski J, Jankowski M, Zdun M, Bukowska D, et al. Gene Ontology Groups and Signaling Pathways Regulating the Process of Avian Satellite Cell Differentiation. Genes. 2022; 13(2):242. https://doi.org/10.3390/genes13020242
Chicago/Turabian StyleGolkar-Narenji, Afsaneh, Paweł Antosik, Shelly Nolin, Marcin Rucinski, Karol Jopek, Agnieszka Zok, Jarosław Sobolewski, Maurycy Jankowski, Maciej Zdun, Dorota Bukowska, and et al. 2022. "Gene Ontology Groups and Signaling Pathways Regulating the Process of Avian Satellite Cell Differentiation" Genes 13, no. 2: 242. https://doi.org/10.3390/genes13020242
APA StyleGolkar-Narenji, A., Antosik, P., Nolin, S., Rucinski, M., Jopek, K., Zok, A., Sobolewski, J., Jankowski, M., Zdun, M., Bukowska, D., Stefańska, K., Jaśkowski, J. M., Piotrowska-Kempisty, H., Mozdziak, P., & Kempisty, B. (2022). Gene Ontology Groups and Signaling Pathways Regulating the Process of Avian Satellite Cell Differentiation. Genes, 13(2), 242. https://doi.org/10.3390/genes13020242