
Microglia Influence Neurofilament Deposition in ALS iPSC-Derived Motor Neurons

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Pluripotent Stem Cells
2.2. Motor Neuron Differentiation and Treatments
2.3. Glial Differentiation and Treatments
2.4. RNA Sequencing
2.5. Western Blot
2.6. Immunocytochemistry
2.7. Multiplex Cytokine Array
2.8. Statistical Analyses
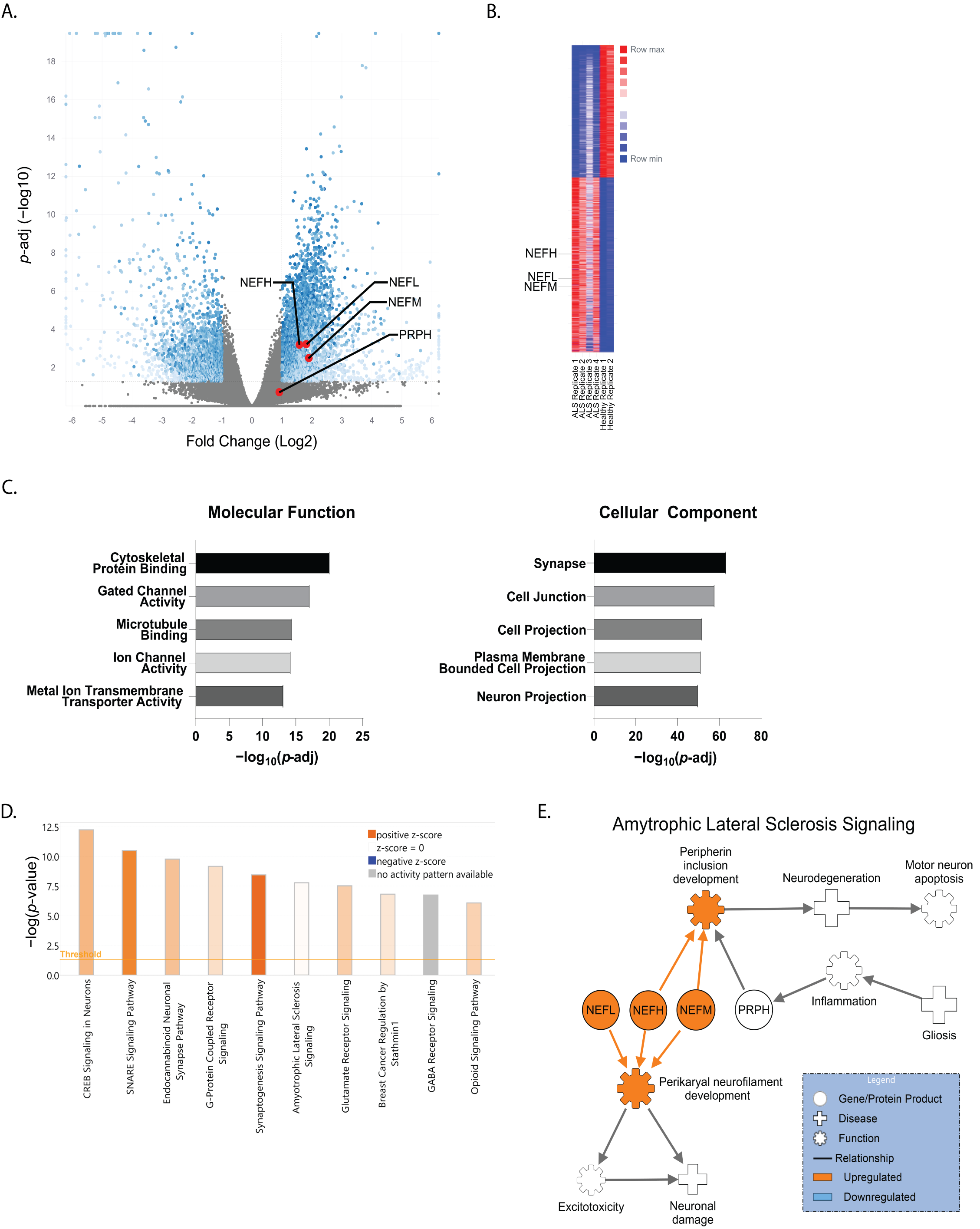
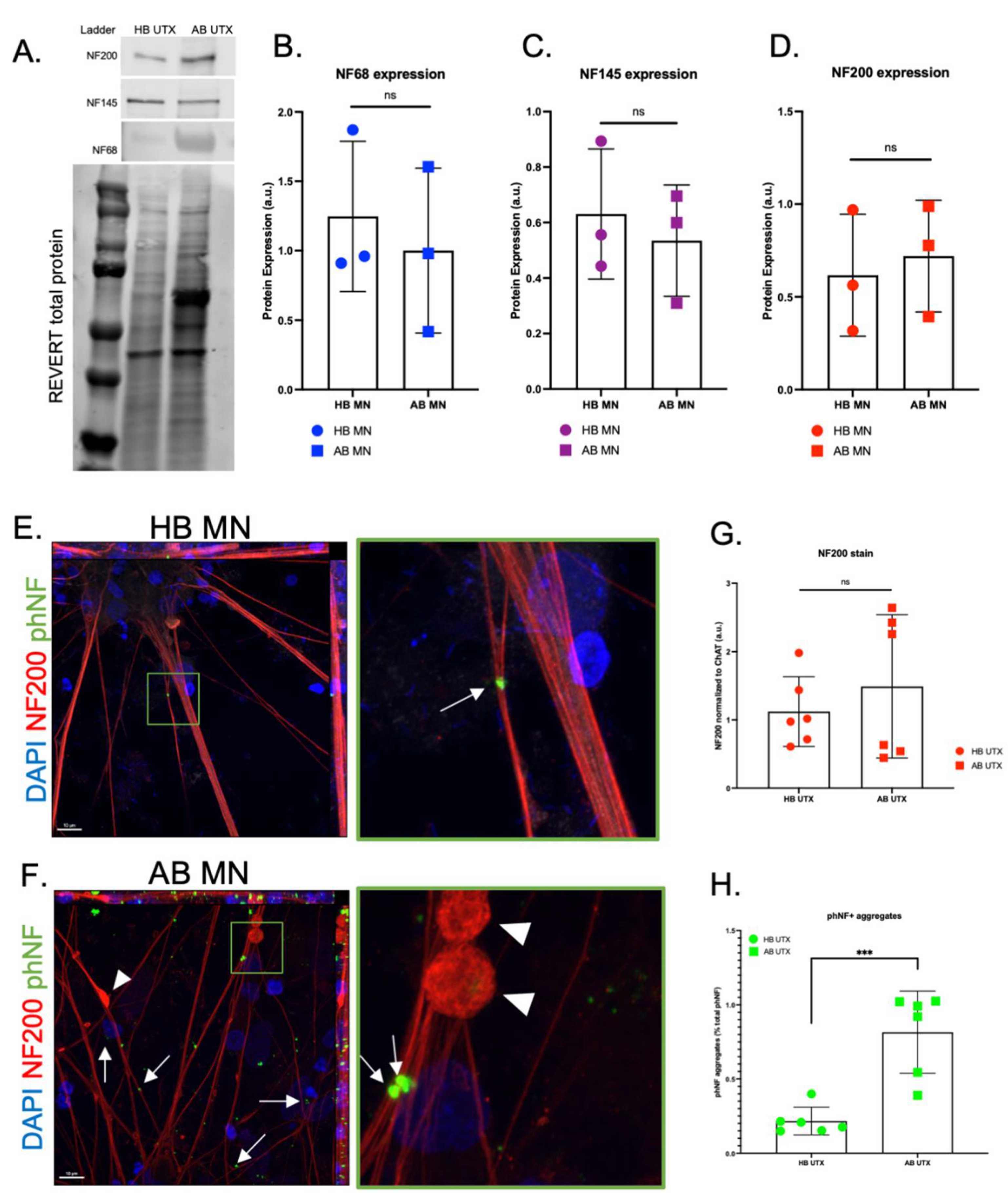
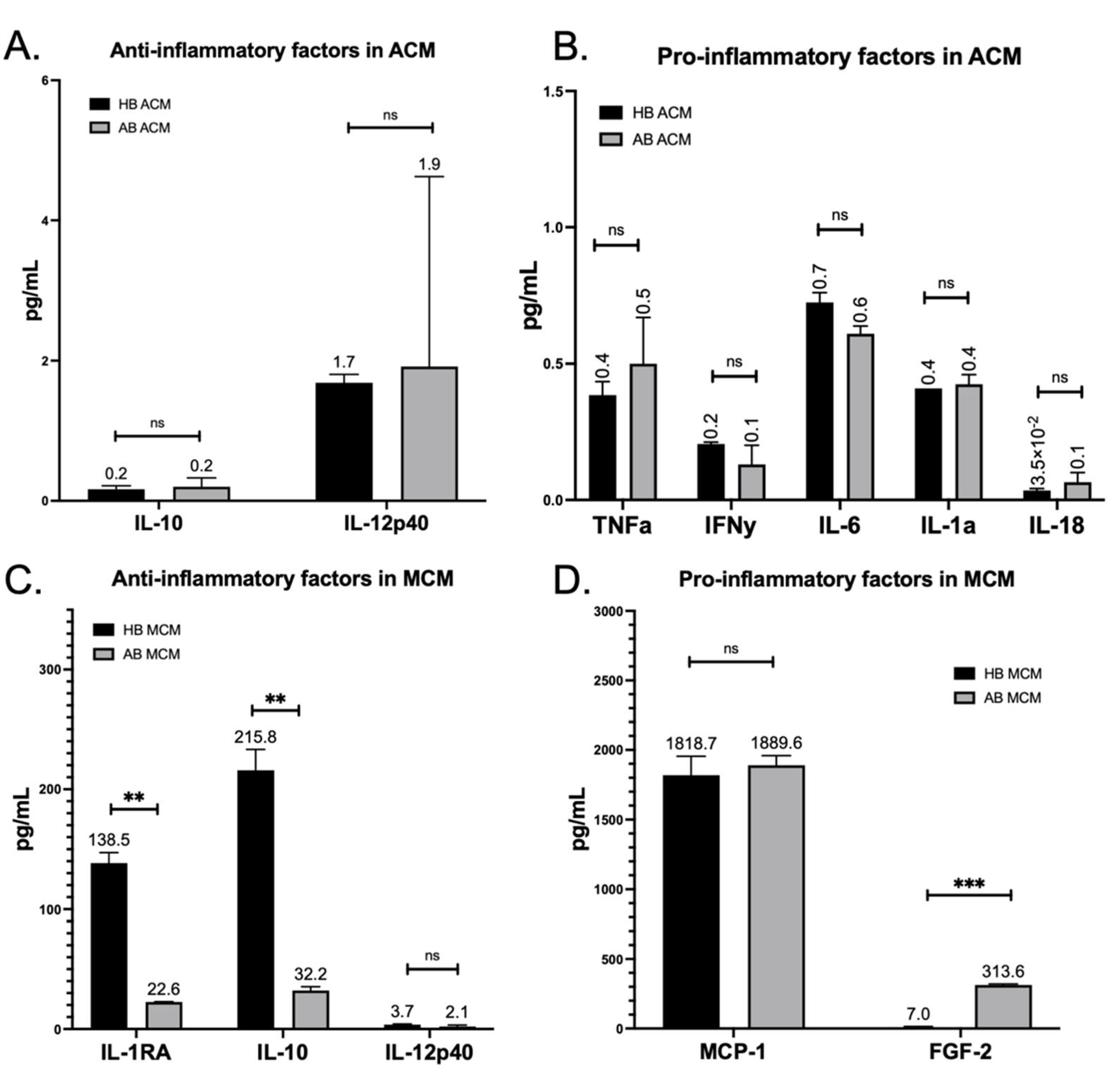
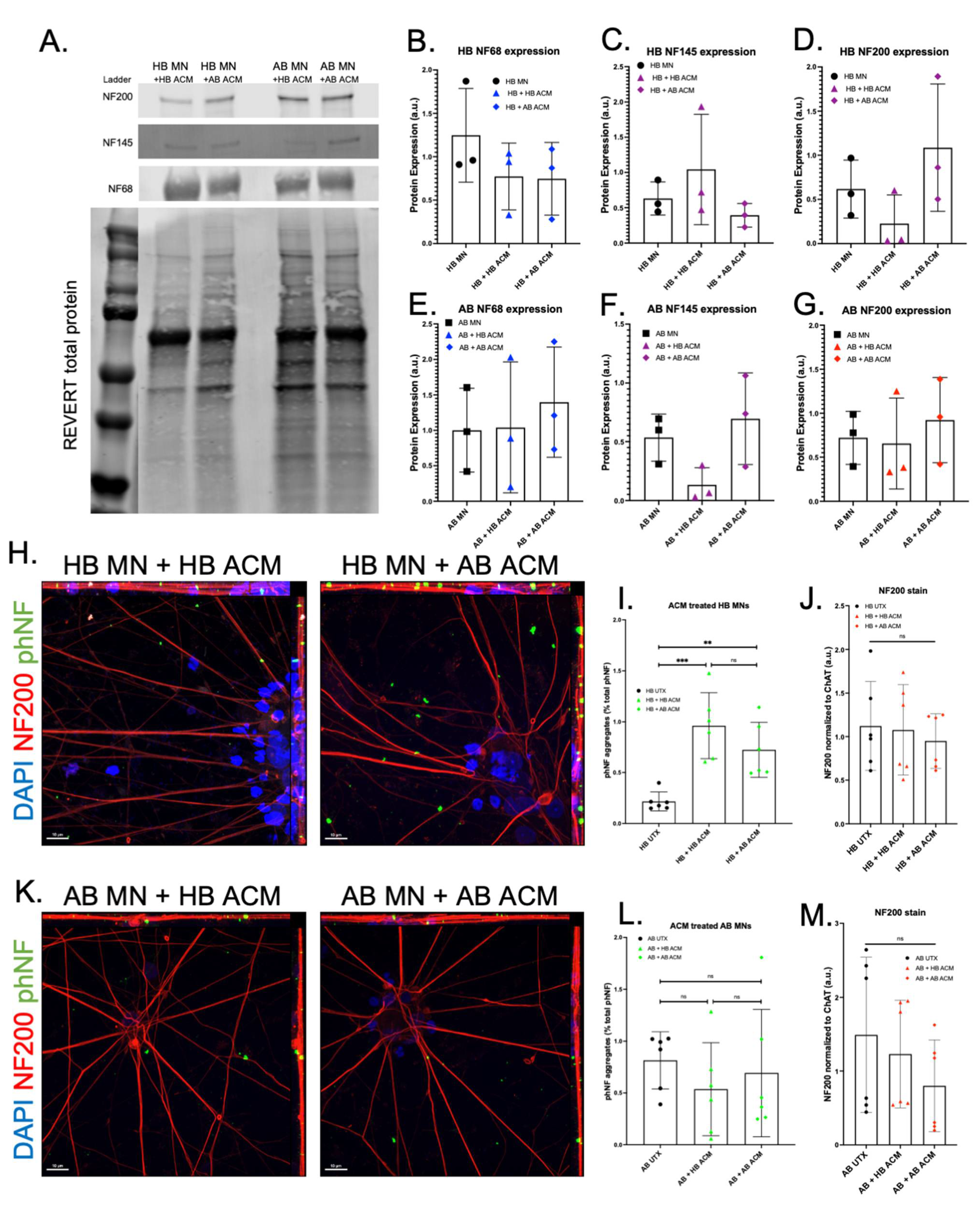
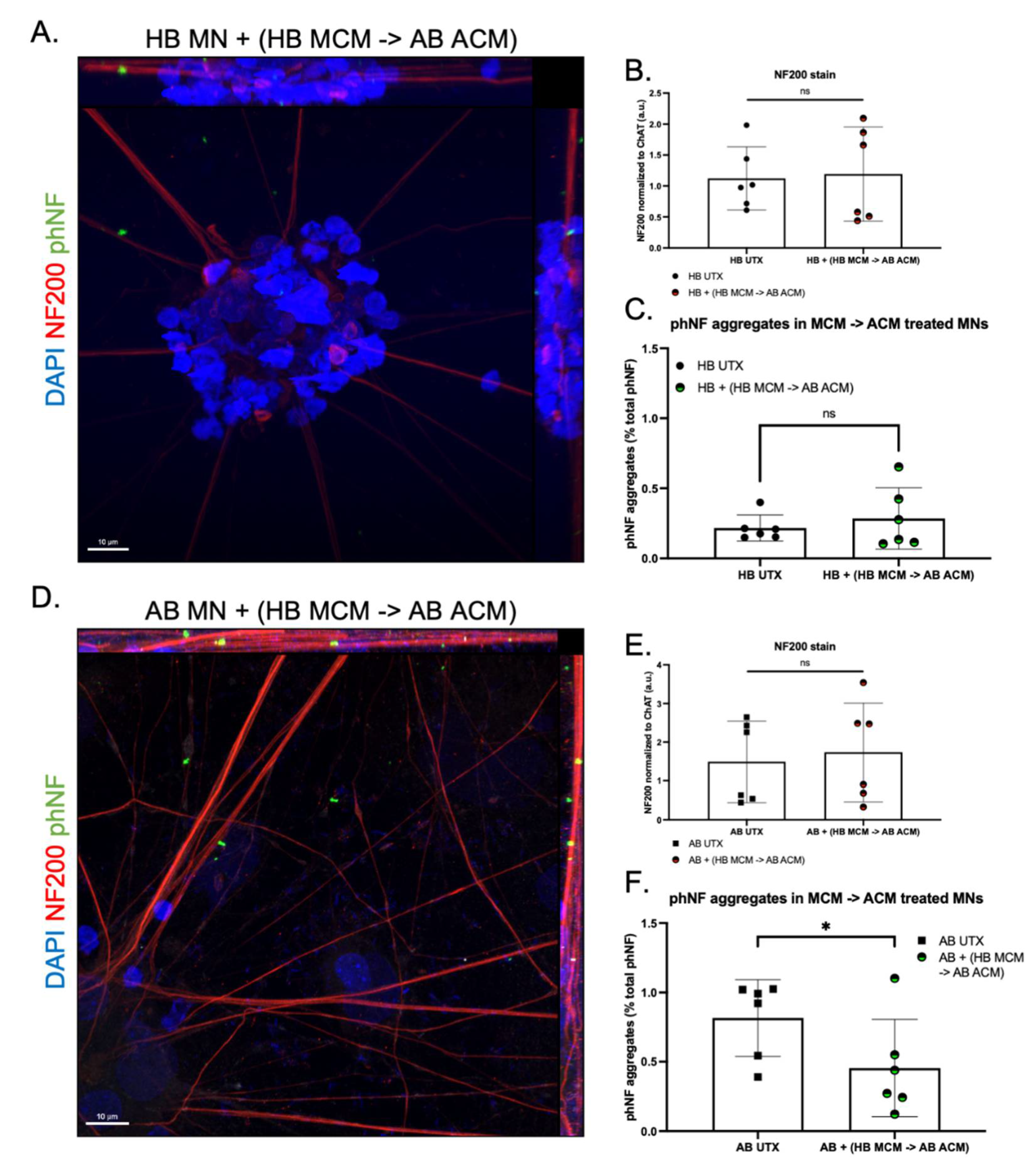
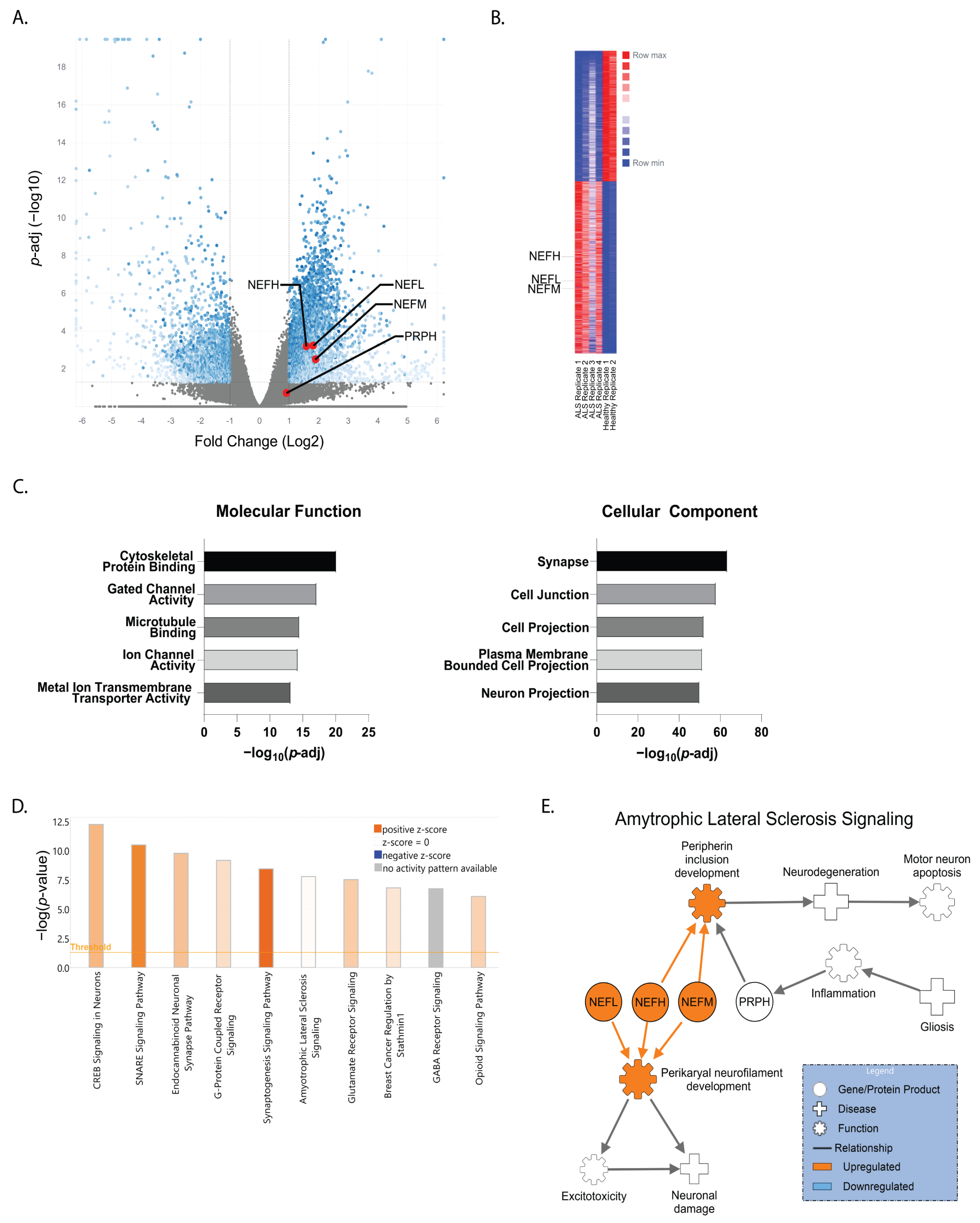
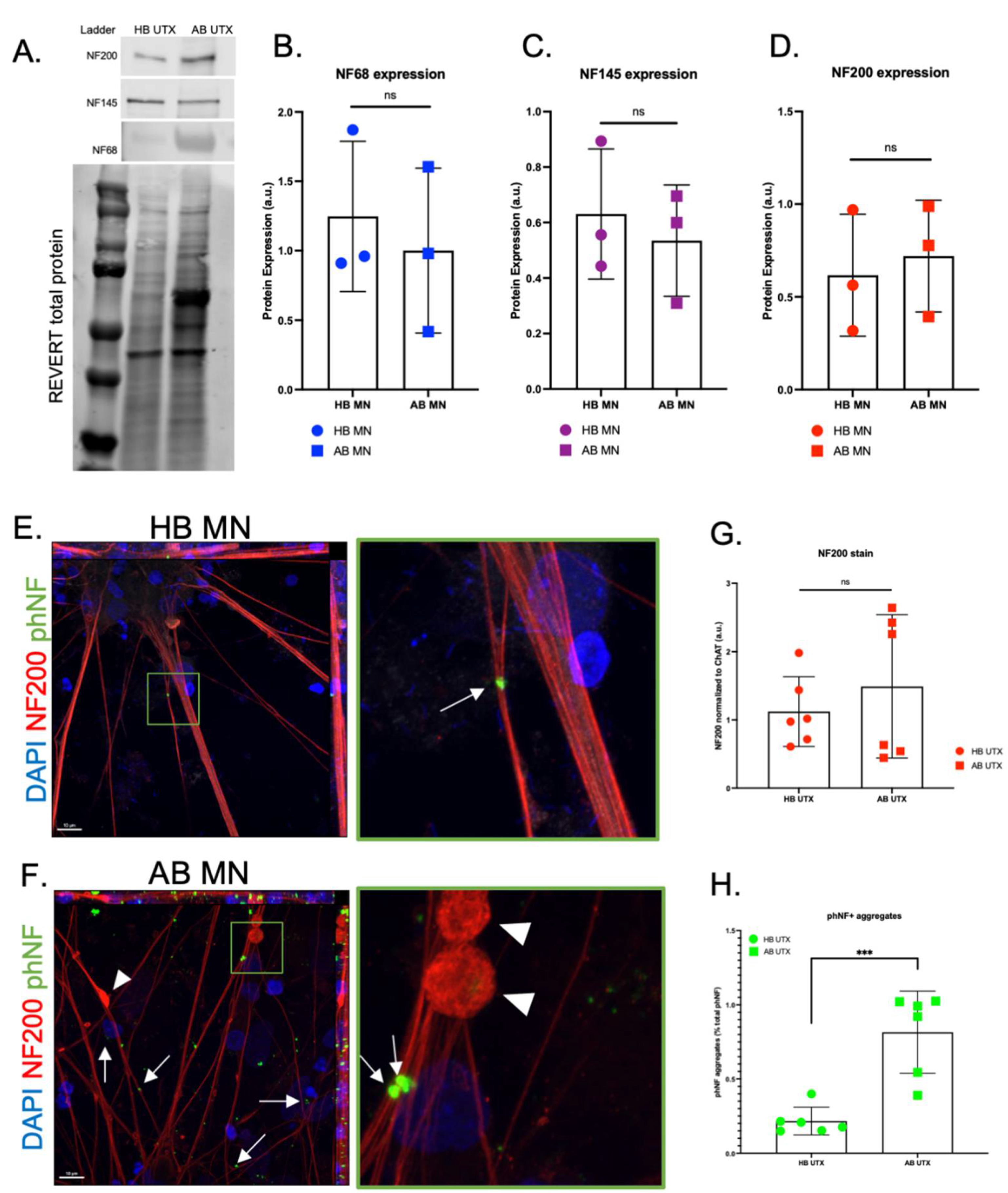
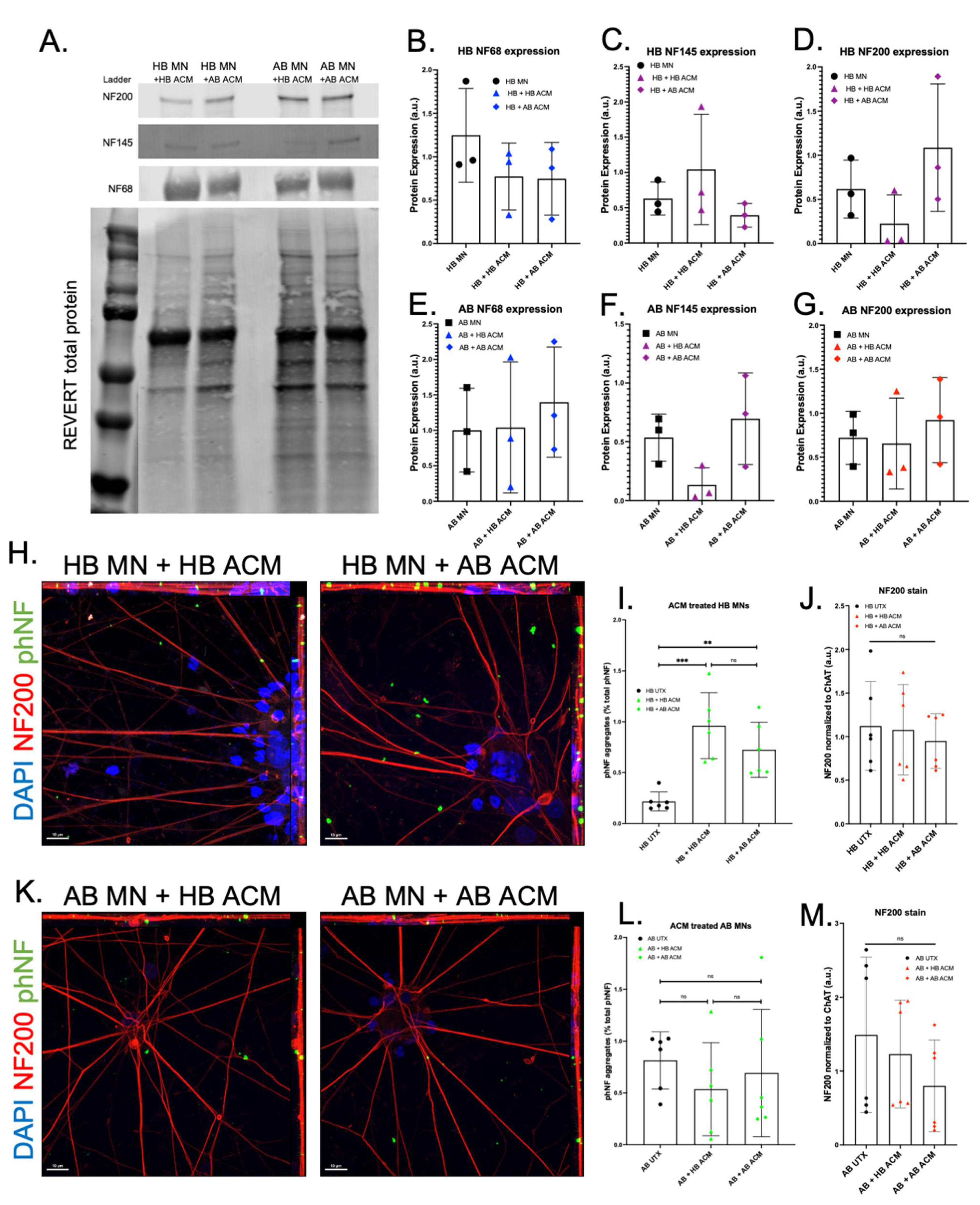
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhasmana, S.; Dhasmana, A.; Narula, A.S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. The panoramic view of amyotrophic lateral sclerosis: A fatal intricate neurological disorder. Life Sci. 2022, 288, 120156. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Amari, M.; Takatama, M.; Aizawa, H.; Mihara, B.; Okamoto, K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2006, 249, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seminary, E.R.; Sison, S.L.; Ebert, A.D. Modeling Protein Aggregation and the Heat Shock Response in ALS iPSC-Derived Motor Neurons. Front. Neurosci. 2018, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figlewicz, D.A.; Krizus, A.; Martinoli, M.-G.; Meininger, V.; Dib, M.; Rouleau, G.A.; Julien, J.-P. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994, 3, 1757–1761. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.; Zoghbi, H. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Ii, K.; Ito, H.; Tanaka, K.; Hirano, A. Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neu-rodegenerative diseases and the elderly. J. Neuropathol. Exp. Neurol. 1997, 56, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-Synuclein Promotes Mitochondrial Deficit and Oxidative Stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Rosengren, L.E.; Karlsson, J.E.; Karlsson, J.O.; Persson, L.I.; Wikkelso, C. Patients with amyotrophic lateral sclerosis and other neu-rodegenerative diseases have increased levels of neurofilament protein in CSF. J. Neurochem. 1996, 67, 2013–2018. [Google Scholar] [CrossRef]
- Zucchi, E.; Bonetto, V.; Sorarù, G.; Martinelli, I.; Parchi, P.; Liguori, R.; Mandrioli, J. Neurofilaments in motor neuron disorders: Towards promising diagnostic and prognostic biomarkers. Mol. Neurodegener. 2020, 15, 58. [Google Scholar] [CrossRef] [PubMed]
- Sferruzza, G.; Bosco, L.; Falzone, Y.M.; Russo, T.; Domi, T.; Quattrini, A.; Filippi, M.; Riva, N. Neurofilament light chain as a biological marker for amyotrophic lateral sclerosis: A meta-analysis study. Amyotroph. Lateral Scler. Front. Degener. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Anderton, B.H.; Ayers, M.; Thorpe, R. Neurofilaments from mammalian central and peripheral nerve share certain polypeptides. FEBS Lett. 1978, 96, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Julien, J.P.; Mushynski, W.E. The distribution of phosphorylation sites among identified proteolytic fragments of mammalian neurofilaments. J. Biol. Chem. 1983, 258, 4019–4025. [Google Scholar] [CrossRef]
- Bocquet, A.; Berges, R.; Frank, R.; Robert, P.; Peterson, A.C.; Eyer, J. Neurofilaments Bind Tubulin and Modulate Its Polymerization. J. Neurosci. 2009, 29, 11043–11054. [Google Scholar] [CrossRef]
- Schwartz, M.L.; Shneidman, P.S.; Bruce, J.; Schlaepfer, W.W. Stabilization of neurofilament transcripts during postnatal development. Brain Res. Mol. Brain Res. 1994, 27, 215–220. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Nguyen, M.D.; Julien, J.-P. Late Onset Death of Motor Neurons in Mice Overexpressing Wild-Type Peripherin. J. Cell Biol. 1999, 147, 531–544. [Google Scholar] [CrossRef]
- Križ, J.; Zhu, Q.; Julien, J.-P.; Padjen, A.L. Electrophysiological properties of axons in mice lacking neurofilament subunit genes: Disparity between conduction velocity and axon diameter in absence of NF-H. Brain Res. 2000, 885, 32–44. [Google Scholar] [CrossRef]
- Sun, X.; Song, J.; Huang, H.; Chen, H.; Qian, K. Modeling hallmark pathology using motor neurons derived from the family and sporadic amyotrophic lateral sclerosis patient-specific iPS cells. Stem Cell Res. Ther. 2018, 9, 315. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.; Huang, C.-L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with iPSCs Reveals that Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Qian, K.; Huang, H.; Peterson, A.; Hu, B.; Maragakis, N.J.; Ming, G.-L.; Chen, H.; Zhang, S.-C. Sporadic ALS Astrocytes Induce Neuronal Degeneration In Vivo. Stem Cell Rep. 2017, 8, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Ackerley, S.; Grierson, A.; Brownlees, J.; Thornhill, P.; Anderton, B.H.; Leigh, P.N.; Shaw, C.E.; Miller, C. Glutamate Slows Axonal Transport of Neurofilaments in Transfected Neurons. J. Cell Biol. 2000, 150, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Julien, J.-P. Amyotrophic Lateral Sclerosis: Unfolding the Toxicity of the Misfolded. Cell 2001, 104, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Lasiene, J.; Yamanaka, K. Glial Cells in Amyotrophic Lateral Sclerosis. Neurol. Res. Int. 2011, 2011, 718987. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Devlin, A.C.; Chouhan, A.K.; Selvaraj, B.T.; Stavrou, M.; Burr, K.; Brivio, V.; He, X.; Mehta, A.R.; Story, D.; et al. Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 2020, 68, 1046–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-Type Nonneuronal Cells Extend Survival of SOD1 Mutant Motor Neurons in ALS Mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef] [Green Version]
- Marchetto, M.C.; Muotri, A.R.; Mu, Y.; Smith, A.M.; Cezar, G.G.; Gage, F.H. Non-Cell-Autonomous Effect of Human SOD1G37R Astrocytes on Motor Neurons Derived from Human Embryonic Stem Cells. Cell Stem Cell 2008, 3, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Benkler, C.; Ben-Zur, T.; Barhum, Y.; Offen, D. Altered astrocytic response to activation in SOD1(G93A) mice and its implications on amyotrophic lateral sclerosis pathogenesis. Glia 2013, 61, 312–326. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiller, K.J.; Restrepo, C.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-Mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.; Cooper, M.A.; Schroder, K.; Yerbury, J.; et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020, 68, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.E.; Patani, R. The microglial component of amyotrophic lateral sclerosis. Brain 2020, 143, 3526–3539. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Seminary, E.R.; Santarriaga, S.; Wheeler, L.; Mejaki, M.; Abrudan, J.; Demos, W.; Zimmermann, M.T.; Urrutia, R.A.; Fee, D.; Barkhaus, P.E.; et al. Motor Neuron Generation from iPSCs from Identical Twins Discordant for Amyotrophic Lateral Sclerosis. Cells 2020, 9, 571. [Google Scholar] [CrossRef] [Green Version]
- Maury, Y.; Côme, J.; Piskorowski, R.A.; Salah-Mohellibi, N.; Chevaleyre, V.; Peschanski, M.; Martinat, C.; Nedelec, S. Combinatorial analysis of developmental cues efficiently converts human plu-ripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 2015, 33, 89–96. [Google Scholar] [CrossRef]
- Allison, R.; Welby, E.; Khayrullina, G.; Burnett, B.; Ebert, A. Viral mediated knockdown of GATA6 in SMA iPSC-derived astrocytes prevents motor neuron loss and microglial activation. bioRxiv 2021. [Google Scholar] [CrossRef]
- McQuade, A.; Coburn, M.; Tu, C.H.; Hasselmann, J.; Davtyan, H.; Blurton-Jones, M. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 2018, 13, 67. [Google Scholar] [CrossRef]
- Kalari, K.R.; Nair, A.A.; Bhavsar, J.D.; O’Brien, D.R.; Davila, J.I.; Bockol, M.A.; Nie, J.; Tang, X.; Baheti, S.; Doughty, J.B.; et al. MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinform. 2014, 15, 224. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarriaga, S.; Luecke, I.; Ebert, A. Detection of soluble and insoluble protein species in patient-derived iPSCs. In Methods in Molecular Biology; Springer-Nature: Berlin/Heidelberg, Germany, 2022; in press. [Google Scholar]
- Liao, Z.; Grimshaw, R.S.; Rosenstreich, D.L. Identification of a specific interleukin 1 inhibitor in the urine of febrile patients. J. Exp. Med. 1984, 159, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Shemer, A.; Scheyltjens, I.; Frumer, G.R.; Kim, J.S.; Grozovski, J.; Ayanaw, S.; Dassa, B.; Van Hove, H.; Chappell-Maor, L.; Boura-Halfon, S.; et al. Interleukin-10 Prevents Pathological Microglia Hyperactivation following Peripheral Endotoxin Challenge. Immunity 2020, 53, 1033–1049.e7. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S. Proximal axonal enlargement in motor neuron disease. Neurology 1968, 18, 841. [Google Scholar] [CrossRef]
- Côté, F.; Collard, J.F.; Julien, J.P. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: A mouse model of amyotrophic lateral sclerosis. Cell 1993, 73, 35–46. [Google Scholar] [CrossRef]
- Xu, Z.; Cork, L.C.; Griffin, J.W.; Cleveland, D.W. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell 1993, 73, 23–33. [Google Scholar] [CrossRef]
- Williamson, T.L.; Bruijn, L.I.; Zhu, Q.; Anderson, K.L.; Anderson, S.D.; Julien, J.-P.; Cleveland, D. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc. Natl. Acad. Sci. USA 1998, 95, 9631–9636. [Google Scholar] [CrossRef] [Green Version]
- Couillard-Despres, S.; Zhu, Q.; Wong, P.C.; Price, D.L.; Cleveland, D.; Julien, J.-P. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc. Natl. Acad. Sci. USA 1998, 95, 9626–9630. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Xu, Z. Overexpression of neurofilament subunit NF-L and NF-H extends survival of a mouse model for amyotrophic lateral sclerosis. Neurosci. Lett. 2000, 281, 72–74. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Hawley, Z.C.E.; Strong, M.J. Dysregulation of human NEFM and NEFH mRNA stability by ALS-linked miRNAs. Mol. Brain 2018, 11, 43. [Google Scholar] [CrossRef]
- Shahheydari, H.; Ragagnin, A.; Walker, A.; Toth, R.P.; Vidal, M.; Jagaraj, C.J.; Perri, E.R.; Konopka, A.; Sultana, J.M.; Atkin, J.D. Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front. Mol. Neurosci. 2017, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Rao, M.; Veeranna; Nixon, R.A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.D.; Bordey, A. The astrocyte odyssey. Prog. Neurobiol. 2008, 86, 342–367. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Y.; Jin, W.-L.; Xu, Y.; Jin, M.-Z. Microglia in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 270–280. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Hong, C.; Chen, M.; Yue, T.; Chen, C.; Wang, Z.; You, Q.; Li, C.; Weng, Q.; et al. ALS-Associated E478G Mutation in Human OPTN (Optineurin) Promotes Inflammation and Induces Neuronal Cell Death. Front. Immunol. 2018, 9, 2647. [Google Scholar] [CrossRef]
- Van Harten, A.C.M.; Phatnani, H.; Przedborski, S. Non-Cell-Autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allison, R.L.; Adelman, J.W.; Abrudan, J.; Urrutia, R.A.; Zimmermann, M.T.; Mathison, A.J.; Ebert, A.D. Microglia Influence Neurofilament Deposition in ALS iPSC-Derived Motor Neurons. Genes 2022, 13, 241. https://doi.org/10.3390/genes13020241
Allison RL, Adelman JW, Abrudan J, Urrutia RA, Zimmermann MT, Mathison AJ, Ebert AD. Microglia Influence Neurofilament Deposition in ALS iPSC-Derived Motor Neurons. Genes. 2022; 13(2):241. https://doi.org/10.3390/genes13020241
Chicago/Turabian StyleAllison, Reilly L., Jacob W. Adelman, Jenica Abrudan, Raul A. Urrutia, Michael T. Zimmermann, Angela J. Mathison, and Allison D. Ebert. 2022. "Microglia Influence Neurofilament Deposition in ALS iPSC-Derived Motor Neurons" Genes 13, no. 2: 241. https://doi.org/10.3390/genes13020241
APA StyleAllison, R. L., Adelman, J. W., Abrudan, J., Urrutia, R. A., Zimmermann, M. T., Mathison, A. J., & Ebert, A. D. (2022). Microglia Influence Neurofilament Deposition in ALS iPSC-Derived Motor Neurons. Genes, 13(2), 241. https://doi.org/10.3390/genes13020241