
Dual Molecular Diagnoses of Recessive Disorders in a Child from Consanguineous Parents: Case Report and Literature Review

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chromosomal Microarray Analysis (CMA)
2.2. Whole Exome Sequencing (WES)
2.3. Sanger Sequencing
2.4. Literature Review
3. Results
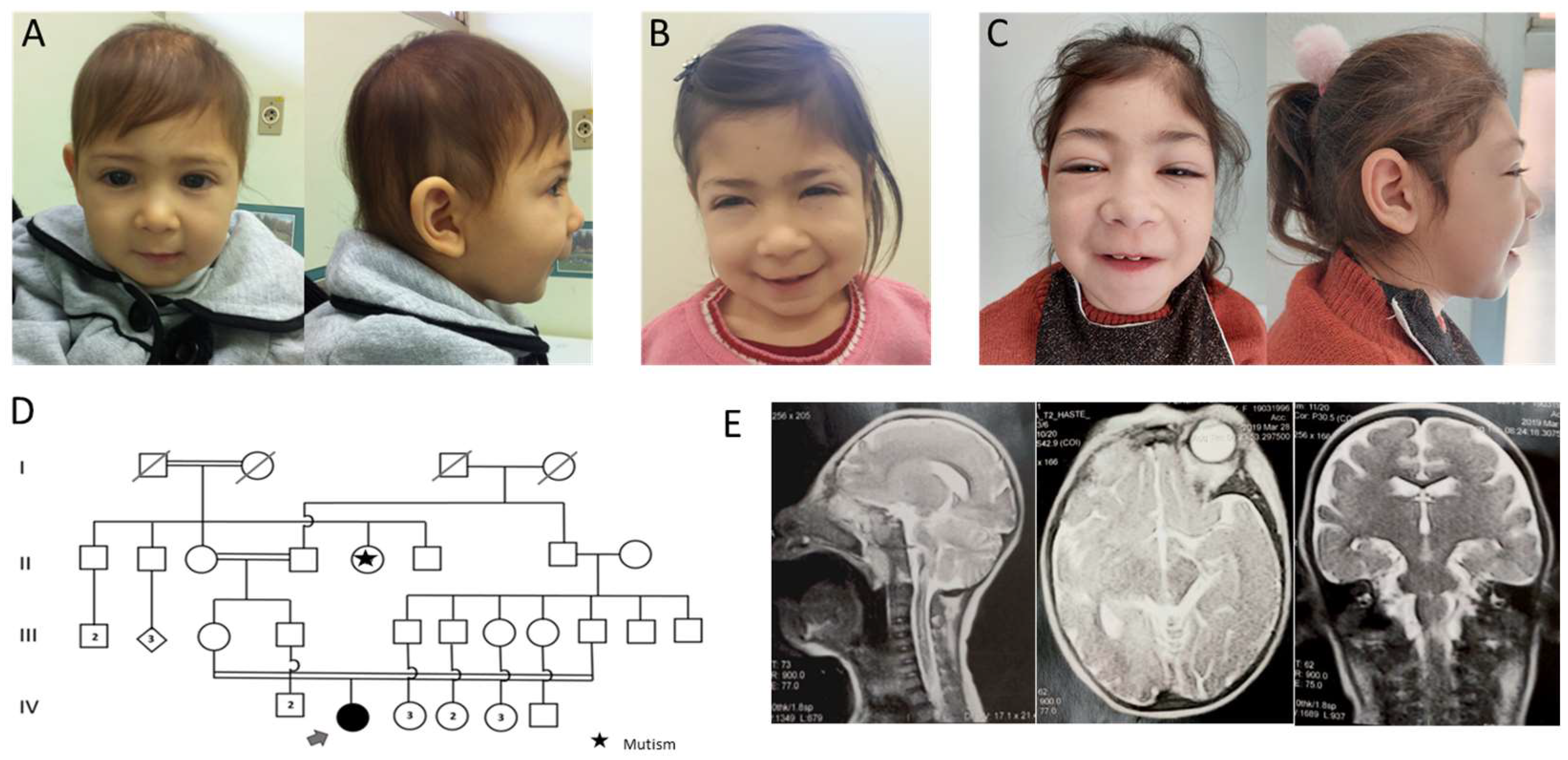
3.1. Case Presentation
3.2. Genetic Tests
3.2.1. CMA
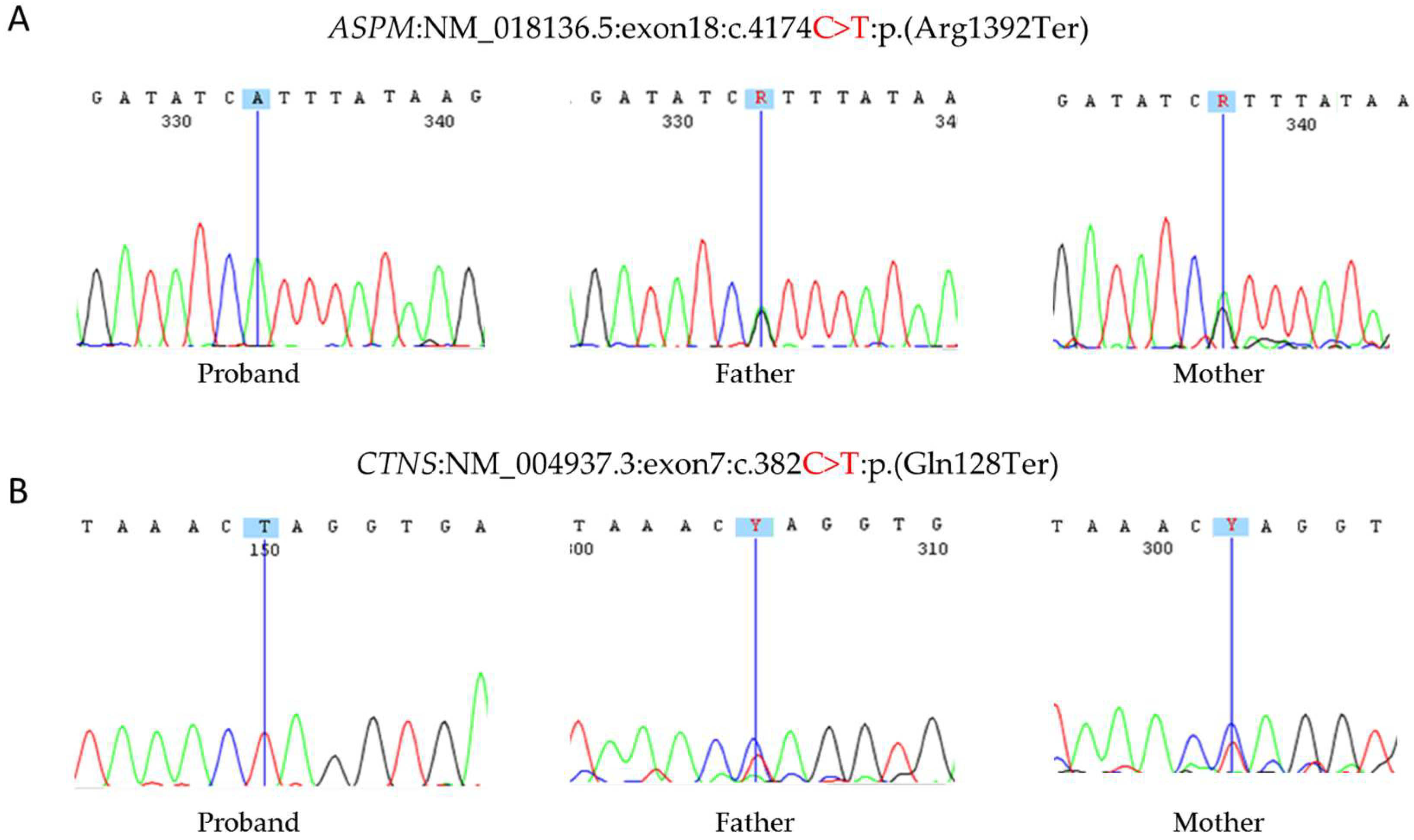
3.2.2. WES
3.2.3. Sanger Sequencing
3.3. Literature Review
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Akdemir, Z.H.C.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Herman, I.; Jolly, A.; Du, H.; Dawood, M.; Abdel-Salam, G.M.H.; Marafi, D.; Mitani, T.; Calame, D.G.; Coban-Akdemir, Z.; Fatih, J.M.; et al. Quantitative dissection of multilocus pathogenic variation in an Egyptian infant with severe neurodevelopmental disorder resulting from multiple molecular diagnoses. Am. J. Med. Genet. A 2022, 188, 735–750. [Google Scholar] [CrossRef]
- Balci, T.; Hartley, T.; Xi, Y.; Dyment, D.; Beaulieu, C.; Bernier, F.; Dupuis, L.; Horvath, G.; Mendoza-Londono, R.; Prasad, C.; et al. Debunking Occam’s razor: Diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin. Genet. 2017, 92, 281–289. [Google Scholar] [CrossRef]
- Smith, E.D.; Blanco, K.; Sajan, S.A.; Hunter, J.M.; Shinde, D.N.; Wayburn, B.; Rossi, M.; Huang, J.; Stevens, C.A.; Muss, C.; et al. A retrospective review of multiple findings in diagnostic exome sequencing: Half are distinct and half are overlapping diagnoses. Genet. Med. 2019, 21, 2199–2207. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef]
- Farwell, K.D.; Shahmirzadi, L.; El-Khechen, D.; Powis, Z.; Chao, E.C.; Davis, B.T.; Baxter, R.M.; Zeng, W.; Mroske, C.; Parra, M.C.; et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 2015, 17, 578–586. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef]
- Karaca, E.; Posey, J.E.; Akdemir, Z.C.; Pehlivan, D.; Harel, T.; Jhangiani, S.N.; Bayram, Y.; Song, X.; Bahrambeigi, V.; Yuregir, O.O.; et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet. Med. 2018, 20, 1528–1537. [Google Scholar] [CrossRef]
- Pehlivan, D.; Bayram, Y.; Gunes, N.; Akdemir, Z.C.; Shukla, A.; Bierhals, T.; Tabakci, B.; Sahin, Y.; Gezdirici, A.; Fatih, J.; et al. The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. Am. J. Hum. Genet. 2019, 105, 132–150. [Google Scholar] [CrossRef]
- Mitani, T.; Isikay, S.; Gezdirici, A.; Gulec, E.Y.; Punetha, J.; Fatih, J.M.; Herman, I.; Akay, G.; Du, H.; Calame, D.G.; et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am. J. Hum. Genet. 2021, 108, 1981–2005. [Google Scholar] [CrossRef]
- de Souza, L.C.; Dos Santos, A.P.; Sgardioli, I.C.; Viguetti-Campos, N.L.; Prota, J.R.M.; De Oliveira-Sobrinho, R.P.; Vieira, T.P.; Gil-Da-Silva-Lopes, V.L. Phenotype comparison among individuals with developmental delay/intellectual disability with or without genomic imbalances. J. Intellect. Disabil. Res. 2019, 63, 1379–1389. [Google Scholar] [CrossRef]
- Correia-Costa, G.R.; Sgardioli, I.C.; dos Santos, A.P.; de Araujo, T.K.; Secolin, R.; Lopes-Cendes, I.; Gil-Da-Silva-Lopes, V.L.; Vieira, T.P. Increased runs of homozygosity in the autosomal genome of Brazilian individuals with neurodevelopmental delay/intellectual disability and/or multiple congenital anomalies investigated by chromosomal microarray analysis. Genet. Mol. Biol. 2022, 45, e20200480. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef]
- Silva, M.; de Leeuw, N.; Mann, K.; Schuring-Blom, H.; Morgan, S.; Giardino, D.; Rack, K.; Hastings, R. European guidelines for constitutional cytogenomic analysis. Eur. J. Hum. Genet. 2019, 27, 1–16. [Google Scholar] [CrossRef]
- Kearney, H.M.; Kearney, J.B.; Conlin, L.K. Diagnostic implications of excessive homozygosity detected by SNP-Based microarrays: Consanguinity, uniparental disomy, and recessive single-gene mutations. Clin. Lab. Med. 2011, 31, 595–613. [Google Scholar] [CrossRef]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.-J.; de Vries, P.; A de Vries, B.B.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Town, M.M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Scliar, M.O.; Yamamoto, G.L.; Wang, J.Y.T.; Zverinova, S.; Karp, T.; Nunes, K.; Ceroni, J.R.M.; de Carvalho, D.L.; da Silva Simões, C.E.; et al. Whole-genome sequencing of 1171 elderly admixed individuals from Brazil. Nat. Commun. 2022, 13, 1004. [Google Scholar] [CrossRef]
- Patrono, C.; Vicia, C.; Giannotti, A.; Bembi, B.; Digilio, M.; Rizzo, C.; Purificato, C.; Martini, C.; Pierini, R.; Santorelli, F. Two novel mutations of the human Δ7-sterol reductase (DHCR7) gene in children with Smith-Lemli-Opitz syndrome. Mol. Cell. Prob. 2002, 16, 315–318. [Google Scholar] [CrossRef]
- Shao, Z.; Masuho, I.; Tumber, A.; Maynes, J.T.; Tavares, E.; Ali, A.; Hewson, S.; Schulze, A.; Kannu, P.; Martemyanov, K.A.; et al. Extended Phenotyping and Functional Validation Facilitate Diagnosis of a Complex Patient Harboring Genetic Variants in MCCC1 and GNB5 Causing Overlapping Phenotypes. Genes 2021, 12, 1352. [Google Scholar] [CrossRef]
- Slachtova, L.; Seda, O.; Behunova, J.; Mistrik, M.; Martasek, P. Genetic and biochemical study of dual hereditary jaundice: Dubin-Johnson and Gilbert’s syndromes. Haplotyping and founder effect of deletion in ABCC2. Eur. J. Hum. Genet. 2016, 24, 704–709. [Google Scholar] [CrossRef]
- Berkun, L.; Slae, M.; Mor-Shaked, H.; Koplewitz, B.; Eventov-Friedman, S.; Harel, T. Homozygous variants in MAPRE2 and CDON in individual with skin folds, growth delay, retinal coloboma, and pyloric stenosis. Am. J. Med. Genet. A 2019, 179, 2454–2458. [Google Scholar] [CrossRef]
- Baltacı, H.N.C.; Taşdelen, E.; Topçu, V.; Eminoğlu, F.T.; Karabulut, H.G. Dual diagnosis of Ochoa syndrome and Niemann-Pick disease type B in a consanguineous family. J. Pediatr. Endocrinol. Metab. 2021, 34, 653–657. [Google Scholar] [CrossRef]
- Cebecauerová, D.; Jirasek, T.; Budisova, L.; Mandys, V.; Volf, V.; Novotna, Z.; Subhanova, I.; Hrebicek, M.; Elleder, M.; Jirsa, M. Dual hereditary jaundice: Simultaneous occurrence of mutations causing Gilbert’s and Dubin-Johnson syndrome. Gastroenterology 2005, 129, 315–320. [Google Scholar] [CrossRef]
- Doğulu, N.; Kose, E.; Kirsaçlioğlu, C.T.; Ezgü, F.S.; Kuloğlu, Z.; Kansu, A.; Eminoglu, F.T. Co-Occurring Atypical Galactosemia and Wilson Disease. Mol. Syndromol. 2022, 13, 454–458. [Google Scholar] [CrossRef]
- Akalın, A.; Şimşek-Kiper, P.Ö.; Taşkıran, E.; Utine, G.E.; Boduroğlu, K. Typical Face, Developmental Delay, and Hearing Loss in a Patient with 3M Syndrome: The Co-Occurrence of Two Rare Conditions. Mol. Syndromol. 2022. [Google Scholar] [CrossRef]
- Al-Dewik, N.; Mohd, H.; Al-Mureikhi, M.; Ali, R.; Al-Mesaifri, F.; Mahmoud, L.; Shahbeck, N.; El-Akouri, K.; Almulla, M.; Al Sulaiman, R.; et al. Clinical exome sequencing in 509 Middle Eastern families with suspected Mendelian diseases: The Qatari experience. Am. J. Med. Genet. A 2019, 179, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Abouelhoda, M.; Assoum, M.; Moghrabi, N.; Rafiullah, R.; Almontashiri, N.; Alowain, M.; Alzaidan, H.; Alsayed, M.; Subhani, S.; et al. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 2019, 104, 1182–1201. [Google Scholar] [CrossRef] [PubMed]
- Pezzani, L.; Pezzoli, L.; Pansa, A.; Facchinetti, B.; Marchetti, D.; Scatigno, A.; Lincesso, A.R.; Perego, L.; Pingue, M.; Pellicioli, I.; et al. Double homozygosity in CEP57 and DYNC2H1 genes detected by WES: Composite or expanded phenotype? Mol. Genet. Genomic. Med. 2020, 8, e1064. [Google Scholar] [CrossRef] [PubMed]
- Rosina, E.; Pezzani, L.; Pezzoli, L.; Marchetti, D.; Bellini, M.; Pilotta, A.; Calabrese, O.; Nicastro, E.; Cirillo, F.; Cereda, A.; et al. Atypical, Composite, or Blended Phenotypes: How Different Molecular Mechanisms Could Associate in Double-Diagnosed Patients. Genes 2022, 13, 1275. [Google Scholar] [CrossRef] [PubMed]
- Valilou, S.F.; Alavi, A.; Pashaei, M.; Firouzabadi, S.G.; Shafeghati, Y.; Nozari, A.; Hadipour, F.; Hadipour, Z.; Estrabadi, B.M.; Noorazar, S.G.; et al. Whole-Exome Sequencing Identifies Three Candidate Homozygous Variants in a Consanguineous Iranian Family with Autism Spectrum Disorder and Skeletal Problems. Mol. Syndromol. 2020, 11, 62–72. [Google Scholar] [CrossRef]
- Umair, M.; Seidel, H.; Ahmed, I.; Ullah, A.; Haack, T.B.; Alhaddad, B.; Jan, A.; Rafique, A.; Strom, T.M.; Ahmad, F.; et al. Ellis-van Creveld syndrome and profound deafness resulted by sequence variants in the EVC/EVC2 and TMC1 genes. J. Genet. 2017, 96, 1005–1014. [Google Scholar] [CrossRef]
- Poot, M. Expanded Phenotypic Spectrum or Multiple Syndromes? Mol. Syndromol. 2022, 13, 361–362. [Google Scholar] [CrossRef]
- Fareed, M.; Afzal, M. Genetics of consanguinity and inbreeding in health and disease. Ann. Hum. Biol. 2017, 44, 99–107. [Google Scholar] [CrossRef]
- Oniya, O.; Neves, K.; Ahmed, B.; Konje, J.C. A review of the reproductive consequences of consanguinity. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 232, 87–96. [Google Scholar] [CrossRef]
- Bond, J.; Roberts, E.; Mochida, G.H.; Hampshire, D.J.; Scott, S.; Askham, J.M.; Springell, K.; Mahadevan, M.; Crow, Y.J.; Markham, A.F.; et al. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 2002, 32, 316–320. [Google Scholar] [CrossRef]
- Bond, J.; Scott, S.; Hampshire, D.J.; Springell, K.; Corry, P.; Abramowicz, M.J.; Mochida, G.H.; Hennekam, R.C.; Maher, E.R.; Fryns, J.-P.; et al. Protein-Truncating Mutations in ASPM Cause Variable Reduction in Brain Size. Am. J. Hum. Genet. 2003, 73, 1170–1177. [Google Scholar] [CrossRef]
- Khan, M.A.; Windpassinger, C.; Ali, M.Z.; Zubair, M.; Gul, H.; Abbas, S.; Khan, S.; Badar, M.; Mohammad, R.M.; Nawaz, Z. Molecular genetic analysis of consanguineous families with primary microcephaly identified pathogenic variants in the ASPM gene. J. Genet. 2017, 96, 383–387. [Google Scholar] [CrossRef]
- Passemard, S.; Verloes, A.; de Villemeur, T.B.; Boespflug-Tanguy, O.; Hernandez, K.; Laurent, M.; Isidor, B.; Alberti, C.; Pouvreau, N.; Drunat, S.; et al. Abnormal spindle-like microcephaly-associated (ASPM) mutations strongly disrupt neocortical structure but spare the hippocampus and long-term memory. Cortex 2016, 74, 158–176. [Google Scholar] [CrossRef]
- Verloes, A.; Drunat, S.; Passemard, S. ASPM Primary Microcephaly. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK555474/ (accessed on 10 March 2022).
- Woods, C.G.; Bond, J.; Enard, W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef]
- Perez, Y.; Bar-Yaacov, R.; Kadir, R.; Wormser, O.; Shelef, I.; Birk, O.; Flusser, H.; Birnbaum, R.Y. Mutations in the microtubule-associated protein MAP11 (C7orf43) cause microcephaly in humans and zebrafish. Brain 2019, 142, 574–585. [Google Scholar] [CrossRef]
- Roberts, E.; Hampshire, D.J.; Pattison, L.; Springell, K.; Jafri, H.; Corry, P.; Mannon, J.; Rashid, Y.; Crow, Y.; Bond, J.; et al. Autosomal recessive primary microcephaly: An analysis of locus heterogeneity and phenotypic variation. J. Med. Genet. 2002, 39, 718–721. [Google Scholar] [CrossRef][Green Version]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephr. 2017, 13, 115–131. [Google Scholar] [CrossRef]
- Elmonem, M.A.; Veys, K.R.; Soliman, N.A.; van Dyck, M.; Heuvel, L.P.V.D.; Levtchenko, E. Cystinosis: A review. Orph. J. Rar. Dis. 2016, 11, 47. [Google Scholar] [CrossRef]
- Saskin, A.; Fulginiti, V.; Birch, A.H.; Trakadis, Y. Prevalence of four Mendelian disorders associated with autism in 2392 affected families. J. Hum. Genet. 2017, 62, 657–659. [Google Scholar] [CrossRef]
- Kars, M.E.; Başak, A.N.; Onat, O.E.; Bilguvar, K.; Choi, J.; Itan, Y.; Çağlar, C.; Palvadeau, R.; Casanova, J.-L.; Cooper, D.N.; et al. The genetic structure of the Turkish population reveals high levels of variation and admixture. Proc. Natl. Acad. Sci. USA 2021, 118, e2026076118. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, T.; Teixeira, A.; Scliar, M.O.; de Barros, J.S.; Lemes, R.B.; Souza, S.; Tolezano, G.; Santos, F.; Tojal, I.; Cypriano, M.; et al. Unraveling the Genetic Architecture of Hepatoblastoma Risk: Birth Defects and Increased Burden of Germline Damaging Variants in Gastrointestinal/Renal Cancer Predisposition and DNA Repair Genes. Front. Genet. 2022, 13, 858396. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Adam, M.P.; Everman, D.B.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Amemiya, A. (Eds.) GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1116/ (accessed on 10 March 2022).
- Mor-Shaked, H.; Rips, J.; Naamat, S.G.; Reich, A.; Elpeleg, O.; Meiner, V.; Harel, T. Parental exome analysis identifies shared carrier status for a second recessive disorder in couples with an affected child. Eur. J. Hum. Genet. 2021, 29, 455–462. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient’s Clinical Findings | ASPM Phenotype * | CTNS Phenotype * |
|---|---|---|
| Growth | ||
| Birth length less than 3rd percentile (HP:0003561) | + | + |
| Failure to thrive in infancy (HP:0001531) | na | + |
| Neuromotor development and neurological features | ||
| Motor delay (HP:0001270) | + | na |
| Delayed speech and language development (HP:0000750) | + | na |
| Intellectual disability, severe (HP:0010864) | + | na |
| Hyperactivity (HP:0000752) | - | na |
| Attention deficit hyperactivity disorder (HP:0007018) | - | na |
| Myopathy (HP:0003198) | na | - |
| Seizure (HP:0001250) | - | na |
| Progressive neurologic deterioration (HP:0002344) | na | + |
| Central Nervous System abnormalities | ||
| Aplasia/Hypoplasia of the corpus callosum (HP:0007370) | + | na |
| Small cerebral cortex (HP:0002472) | + | na |
| Lissencephaly (HP:0001339) | + | na |
| Hypoplasia of the pons (HP:0012110) | - | na |
| Hypoplasia of the frontal lobes (HP:0007333) | + | na |
| Aplasia/Hypoplasia of the cerebellum (HP:00007360) | + | na |
| Ventriculomegaly (HP:0002119) | - | na |
| Cerebral atrophy (HP:0002059) | na | + |
| Head and neck | ||
| Primary microcephaly (HP:0011451) | + | na |
| Sloping forehead (HP:0000340) | + | + |
| Narrow forehead (HP:0000341) | - | na |
| Proptosis (HP:0000520) | - | na |
| Highly arched eyebrows (HP:0002553) | - | na |
| Ears | ||
| Hearing impairment (HP:0000365) | - | na |
| Eyes | ||
| Photophobia (HP:0000613) | na | + |
| Peripheral retinal degeneration (HP:0007769) | na | + |
| Visual loss (HP:0000572) | na | + |
| Corneal crystals (HP:0000531) | na | + |
| Voice | ||
| Weak voice (HP:0001621) | na | - |
| Skeletal | ||
| Delayed skeletal maturation (HP:0002750) | na | + |
| Genu valgum (HP:0002857) | na | + |
| Digestive system | ||
| Hepatomegaly (HP:0002240) | na | + |
| Urinary system | ||
| Renal Fanconi syndrome (HP:0001994) | na | + |
| Stage 5 chronic kidney disease (HP:0003774) | na | + |
| Nephrolithiasis (HP:00000787) | na | nr |
| Hypophosphatemic rickets (HP:0004912) | na | + |
| Episodic metabolic acidosis (HP:0004911) | na | + |
| Polyuria (HP:0000103) | na | + |
| Generalized aminoaciduria (HP:0002909) | na | + |
| Renal Hypophosphatemia (HP:0008732) | na | nr |
| Hyponatremia (HP:00029202) | na | + |
| Microscopic hematuria (HP:0002907) | na | + |
| Endocrine System | ||
| Primary hypothyroidism (HP:0000832) | na | + |
| Exocrine pancreatic insufficiency (HP:0001738) | na | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia-Costa, G.R.; dos Santos, A.M.; de Leeuw, N.; Rigatto, S.Z.P.; Belangero, V.M.S.; Steiner, C.E.; Gil-da-Silva-Lopes, V.L.; Vieira, T.P. Dual Molecular Diagnoses of Recessive Disorders in a Child from Consanguineous Parents: Case Report and Literature Review. Genes 2022, 13, 2377. https://doi.org/10.3390/genes13122377
Correia-Costa GR, dos Santos AM, de Leeuw N, Rigatto SZP, Belangero VMS, Steiner CE, Gil-da-Silva-Lopes VL, Vieira TP. Dual Molecular Diagnoses of Recessive Disorders in a Child from Consanguineous Parents: Case Report and Literature Review. Genes. 2022; 13(12):2377. https://doi.org/10.3390/genes13122377
Chicago/Turabian StyleCorreia-Costa, Gabriela Roldão, Ana Mondadori dos Santos, Nicole de Leeuw, Sumara Zuanazi Pinto Rigatto, Vera Maria Santoro Belangero, Carlos Eduardo Steiner, Vera Lúcia Gil-da-Silva-Lopes, and Társis Paiva Vieira. 2022. "Dual Molecular Diagnoses of Recessive Disorders in a Child from Consanguineous Parents: Case Report and Literature Review" Genes 13, no. 12: 2377. https://doi.org/10.3390/genes13122377
APA StyleCorreia-Costa, G. R., dos Santos, A. M., de Leeuw, N., Rigatto, S. Z. P., Belangero, V. M. S., Steiner, C. E., Gil-da-Silva-Lopes, V. L., & Vieira, T. P. (2022). Dual Molecular Diagnoses of Recessive Disorders in a Child from Consanguineous Parents: Case Report and Literature Review. Genes, 13(12), 2377. https://doi.org/10.3390/genes13122377