




Investigating the Diversity of Tuberculosis Spoligotypes with Dimensionality Reduction and Graph Theory


Abstract
1. Introduction
2. Spolmap: Enriching the Visualization of CRISPR Diversity
3. Spolgraph: Adding a Graph Structure to the Spolmap Representation
3.1. Biological Reasons to Be Interested in a Graph Structure
3.2. Introducing the Graph Structure


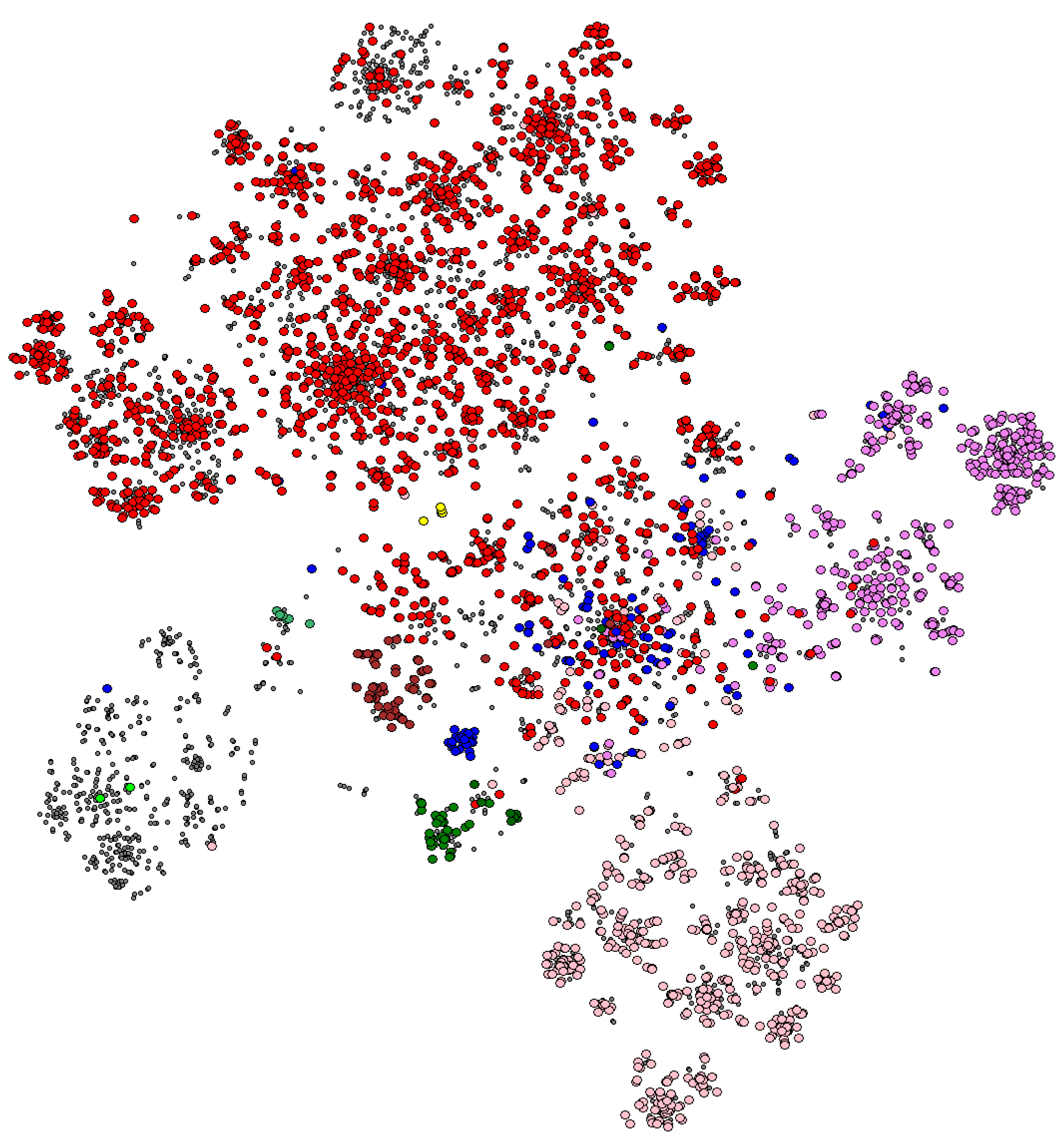
3.3. Investigating the SpolGraph
3.3.1. The Lineages in the Graph
3.3.2. The Connectivity of the Graph
3.3.3. In-Depth Study of Certain Aspects of the Graph
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Refrégier, G.; Sola, C.; Guyeux, C. Unexpected diversity of CRISPR unveils some evolutionary patterns of repeated sequences in Mycobacterium tuberculosis. BMC Genom. 2020, 21, 841. [Google Scholar] [CrossRef] [PubMed]
- Groenen, P.M.; Bunschoten, A.E.; Soolingen, D.V.; Errtbden, J.D.V. Nature of DNA polymorphism in the direct repeat cluster of Mycobacterium tuberculosis; application for strain differentiation by a novel typing method. Mol. Microbiol. 1993, 10, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Kamerbeek, J.; Schouls, L.; Kolk, A.; Van Agterveld, M.; Van Soolingen, D.; Kuijper, S.; Bunschoten, A.; Molhuizen, H.; Shaw, R.; Goyal, M.; et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 1997, 35, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Brudey, K.; Driscoll, J.R.; Rigouts, L.; Prodinger, W.M.; Gori, A.; Al-Hajoj, S.A.; Allix, C.; Aristimuño, L.; Arora, J.; Baumanis, V.; et al. Mycobacterium tuberculosis complex genetic diversity: Mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Kato-Maeda, M.; Gagneux, S.; Flores, L.; Kim, E.; Small, P.; Desmond, E.; Hopewell, P. Strain classification of Mycobacterium tuberculosis: Congruence between large sequence polymorphisms and spoligotypes. Int. J. Tuberc. Lung Dis. 2011, 15, 131–133. [Google Scholar] [PubMed]
- Coll, F.; McNerney, R.; Guerra-Assunção, J.A.; Glynn, J.R.; Perdigão, J.; Viveiros, M.; Portugal, I.; Pain, A.; Martin, N.; Clark, T.G. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 2014, 5, 4812. [Google Scholar] [CrossRef] [PubMed]
- Stucki, D.; Brites, D.; Jeljeli, L.; Coscolla, M.; Liu, Q.; Trauner, A.; Fenner, L.; Rutaihwa, L.; Borrell, S.; Luo, T.; et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. 2016, 48, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Palittapongarnpim, P.; Ajawatanawong, P.; Viratyosin, W.; Smittipat, N.; Disratthakit, A.; Mahasirimongkol, S.; Yanai, H.; Yamada, N.; Nedsuwan, S.; Imasanguan, W.; et al. Evidence for host-bacterial co-evolution via genome sequence analysis of 480 Thai Mycobacterium tuberculosis lineage 1 isolates. Sci. Rep. 2018, 8, 11597. [Google Scholar] [CrossRef] [PubMed]
- Shitikov, E.; Kolchenko, S.; Mokrousov, I.; Bespyatykh, J.; Ischenko, D.; Ilina, E.; Govorun, V. Evolutionary pathway analysis and unified classification of East Asian lineage of Mycobacterium tuberculosis. Sci. Rep. 2017, 7, 9227. [Google Scholar] [CrossRef] [PubMed]
- Guyeux, C.; Senelle, G.; Refrégier, G.; Bretelle-Establet, F.; Cambau, E.; Sola, C. Connection between two historical tuberculosis outbreak sites in Japan, Honshu, by a new ancestral Mycobacterium tuberculosis L2 sublineage. Epidemiol. Infect. 2022, 150, e56. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Classification and nomenclature of CRISPR-Cas systems: Where from here? CRISPR J. 2018, 1, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Freidlin, P.J.; Nissan, I.; Luria, A.; Goldblatt, D.; Schaffer, L.; Kaidar-Shwartz, H.; Chemtob, D.; Dveyrin, Z.; Head, S.R.; Rorman, E. Structure and variation of CRISPR and CRISPR-flanking regions in deleted-direct repeat region Mycobacterium tuberculosis complex strains. BMC Genom. 2017, 18, 168. [Google Scholar] [CrossRef] [PubMed]
- Tsolaki, A.G.; Hirsh, A.E.; DeRiemer, K.; Enciso, J.A.; Wong, M.Z.; Hannan, M.; de la Salmoniere, Y.O.L.G.; Aman, K.; Kato-Maeda, M.; Small, P.M. Functional and evolutionary genomics of Mycobacterium tuberculosis: Insights from genomic deletions in 100 strains. Proc. Natl. Acad. Sci. USA 2004, 101, 4865–4870. [Google Scholar] [CrossRef] [PubMed]
- Van Embden, J.; Van Gorkom, T.; Kremer, K.; Jansen, R.; Van der Zeijst, B.; Schouls, L. Genetic variation and evolutionary origin of the direct repeat locus of Mycobacterium tuberculosis complex bacteria. J. Bacteriol. 2000, 182, 2393–2401. [Google Scholar] [CrossRef] [PubMed]
- Coll, F.; Mallard, K.; Preston, M.D.; Bentley, S.; Parkhill, J.; McNerney, R.; Martin, N.; Clark, T.G. SpolPred: Rapid and accurate prediction of Mycobacterium tuberculosis spoligotypes from short genomic sequences. Bioinformatics 2012, 28, 2991–2993. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.; Teo, Y.Y.; Ong, R.T.H. SpoTyping: Fast and accurate in silico Mycobacterium spoligotyping from sequence reads. Genome Med. 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Guyeux, C.; Sola, C.; Noûs, C.; Refrégier, G. CRISPRbuilder-TB: “CRISPR-builder for tuberculosis”. Exhaustive reconstruction of the CRISPR locus in mycobacterium tuberculosis complex using SRA. PLoS Comput. Biol. 2021, 17, e1008500. [Google Scholar] [CrossRef] [PubMed]
- Guyeux, C.; Refrégier, G.; Sola, C. Spolmap: An enriched visualization of CRISPR diversity. In Proceedings of the 9th International Work-Conference on Bioinformatics and Biomedical Engineering, Gran Canaria, Spain, 27–30 June 2022; Lncs, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2022; Volume 13347, pp. 300–308. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Guyeux, C.; Al-Nuaimi, B.; AlKindy, B.; Couchot, J.F.; Salomon, M. On the reconstruction of the ancestral bacterial genomes in genus Mycobacterium and Brucella. BMC Syst. Biol. Iwbbio 2017 Spec. Issue 2018, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; David, A.; Zozio, T.; Rastogi, N. Macro-geographical specificities of the prevailing tuberculosis epidemic as seen through SITVIT2, an updated version of the Mycobacterium tuberculosis genotyping database. Infect. Genet. Evol. 2019, 72, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, G.; Drake, F.L., Jr. Python Reference Manual; Centrum voor Wiskunde en Informatica: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring Network Structure, Dynamics, and Function using NetworkX. In Proceedings of the 7th Python in Science Conference, Pasadena, CA, USA, 19–24 August 2008; pp. 11–15. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Lineage | Number of Nodes |
|---|---|
| 1 | 1415 |
| 2 | 314 |
| 3 | 970 |
| 4 | 5449 |
| 5 | 98 |
| 6 | 120 |
| 7 | 9 |
| Lineage | Mean | Standard Deviation |
|---|---|---|
| 1 | 4.60 | 1.50 |
| 2 | 3.06 | 1.77 |
| 3 | 3.68 | 1.35 |
| 4 | 4.07 | 1.65 |
| 5 | 3.93 | 1.30 |
| 6 | 3.47 | 1.08 |
| 7 | 2.89 | 0.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senelle, G.; Guyeux, C.; Refrégier, G.; Sola, C. Investigating the Diversity of Tuberculosis Spoligotypes with Dimensionality Reduction and Graph Theory. Genes 2022, 13, 2328. https://doi.org/10.3390/genes13122328
Senelle G, Guyeux C, Refrégier G, Sola C. Investigating the Diversity of Tuberculosis Spoligotypes with Dimensionality Reduction and Graph Theory. Genes. 2022; 13(12):2328. https://doi.org/10.3390/genes13122328
Chicago/Turabian StyleSenelle, Gaetan, Christophe Guyeux, Guislaine Refrégier, and Christophe Sola. 2022. "Investigating the Diversity of Tuberculosis Spoligotypes with Dimensionality Reduction and Graph Theory" Genes 13, no. 12: 2328. https://doi.org/10.3390/genes13122328
APA StyleSenelle, G., Guyeux, C., Refrégier, G., & Sola, C. (2022). Investigating the Diversity of Tuberculosis Spoligotypes with Dimensionality Reduction and Graph Theory. Genes, 13(12), 2328. https://doi.org/10.3390/genes13122328