
Genetic Variants at the Nebulette Locus Are Associated with Myxomatous Mitral Valve Disease Severity in Cavalier King Charles Spaniels

Abstract
1. Introduction
2. Materials and Methods
2.1. Blood Sampling and Echocardiographic Assessment
2.2. DNA Extraction and Array-Based Genotyping
2.3. Allele Frequencies for Risk Variants across Dogs
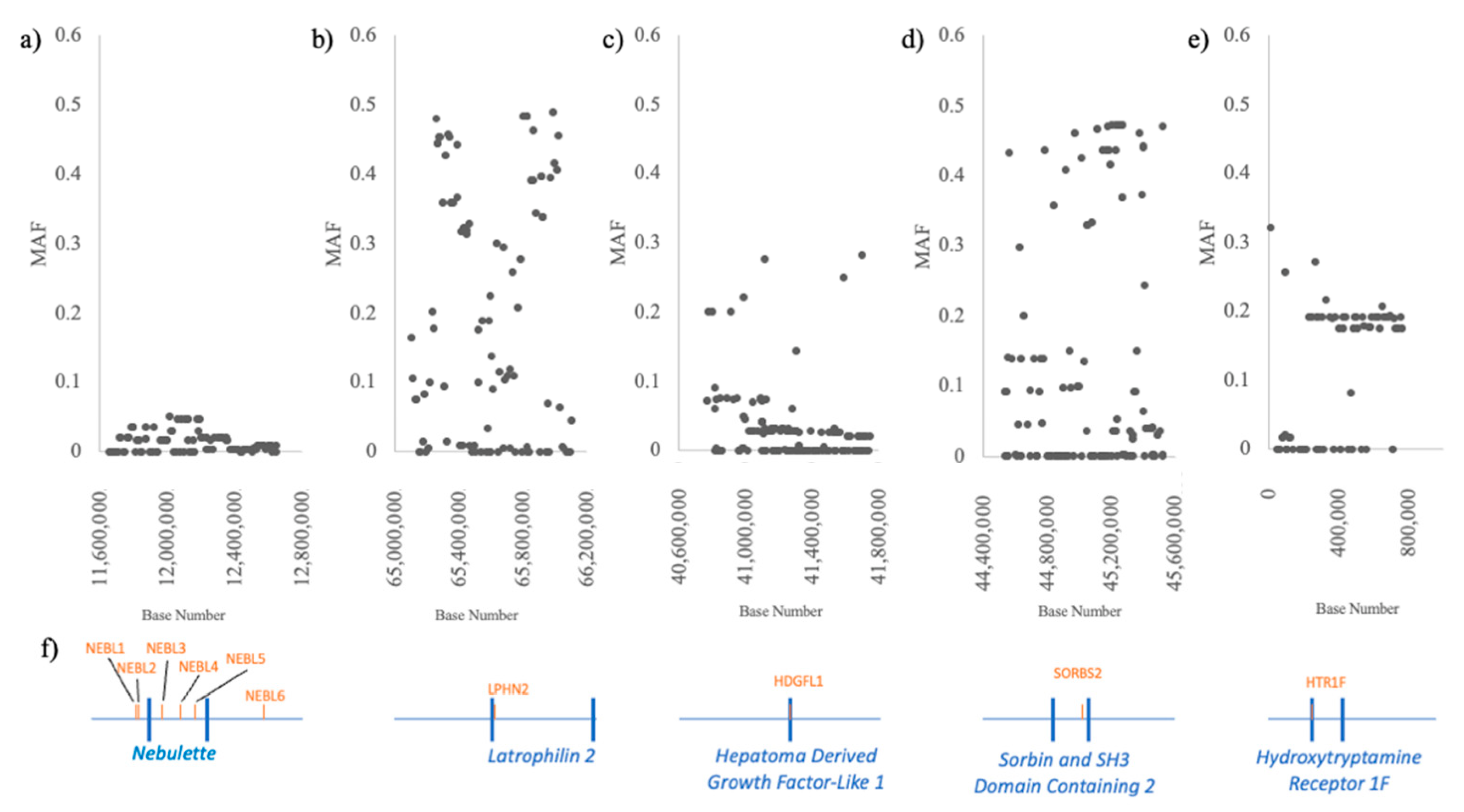
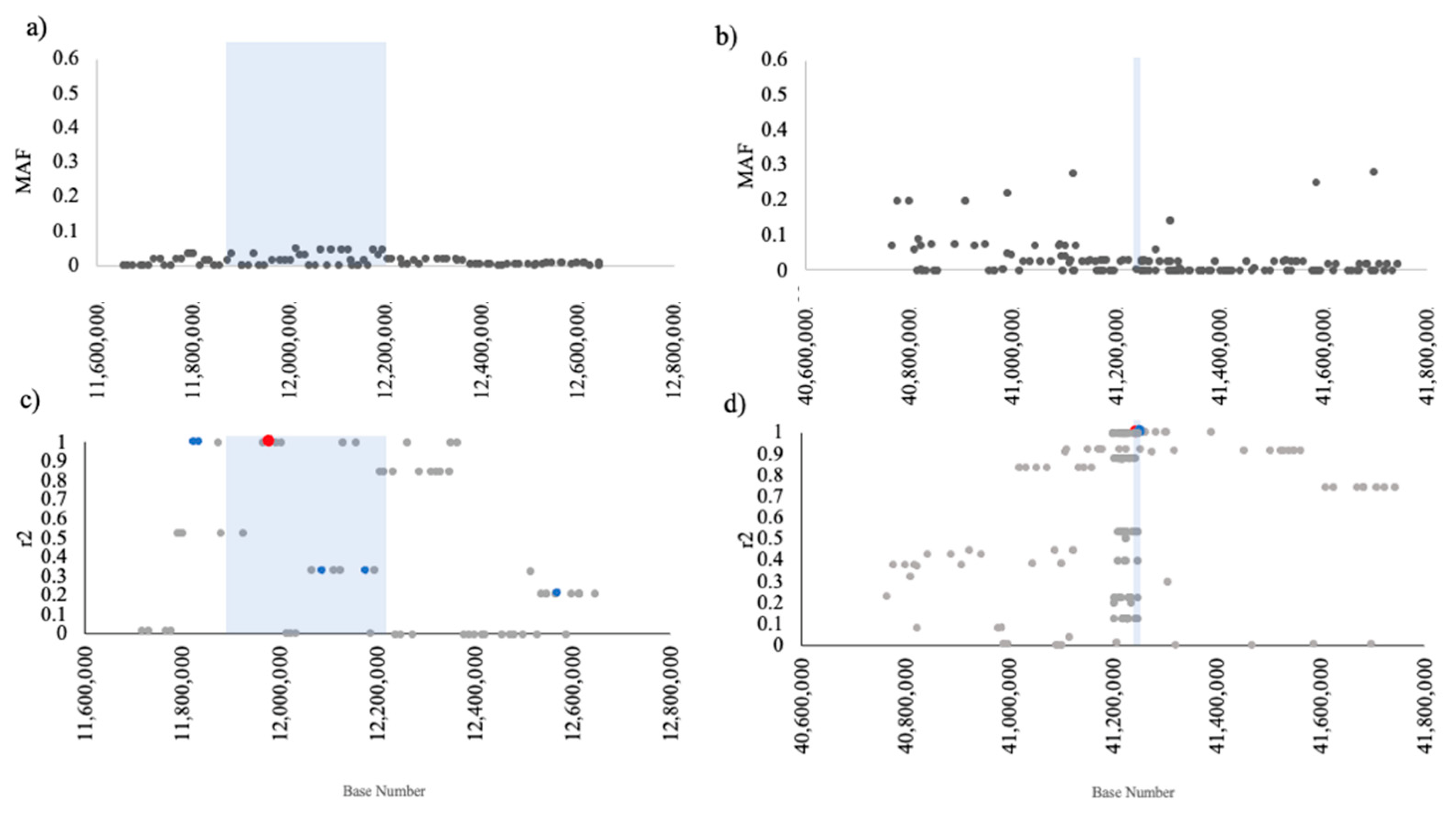
2.4. Regional Analysis of Array Genotypes
2.5. Analysis of Risk Variants
2.6. Analysis of Physiological Data
3. Results
3.1. Phenotypic and Echocardiographic Measurements
3.2. DNA Extraction and Array-Based Genotyping
3.3. Allele Frequencies for Risk Variants across Dog Breeds
3.4. Regional Analysis of Array Genotypes
3.5. Identified Disease-Risk Variants
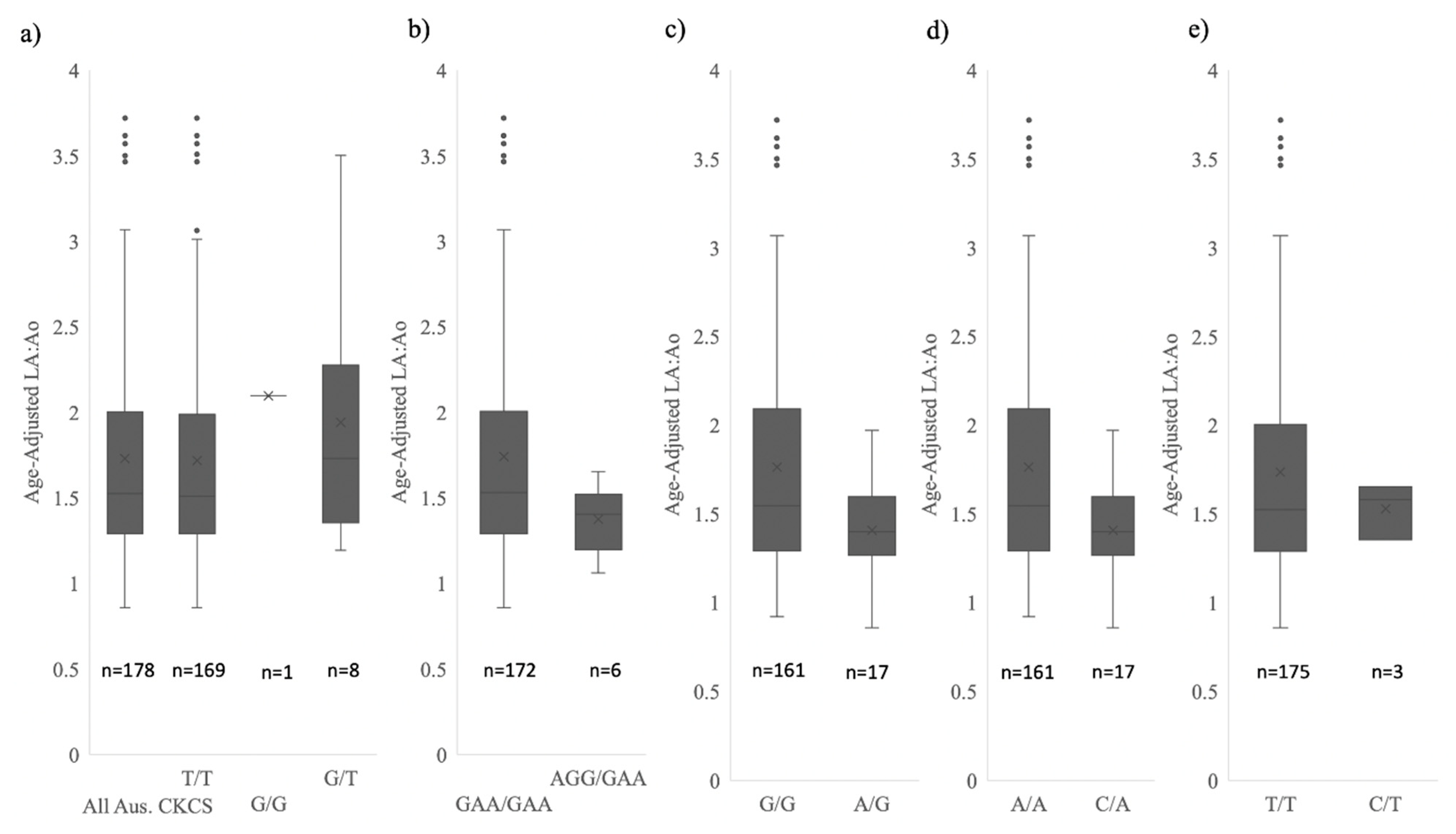
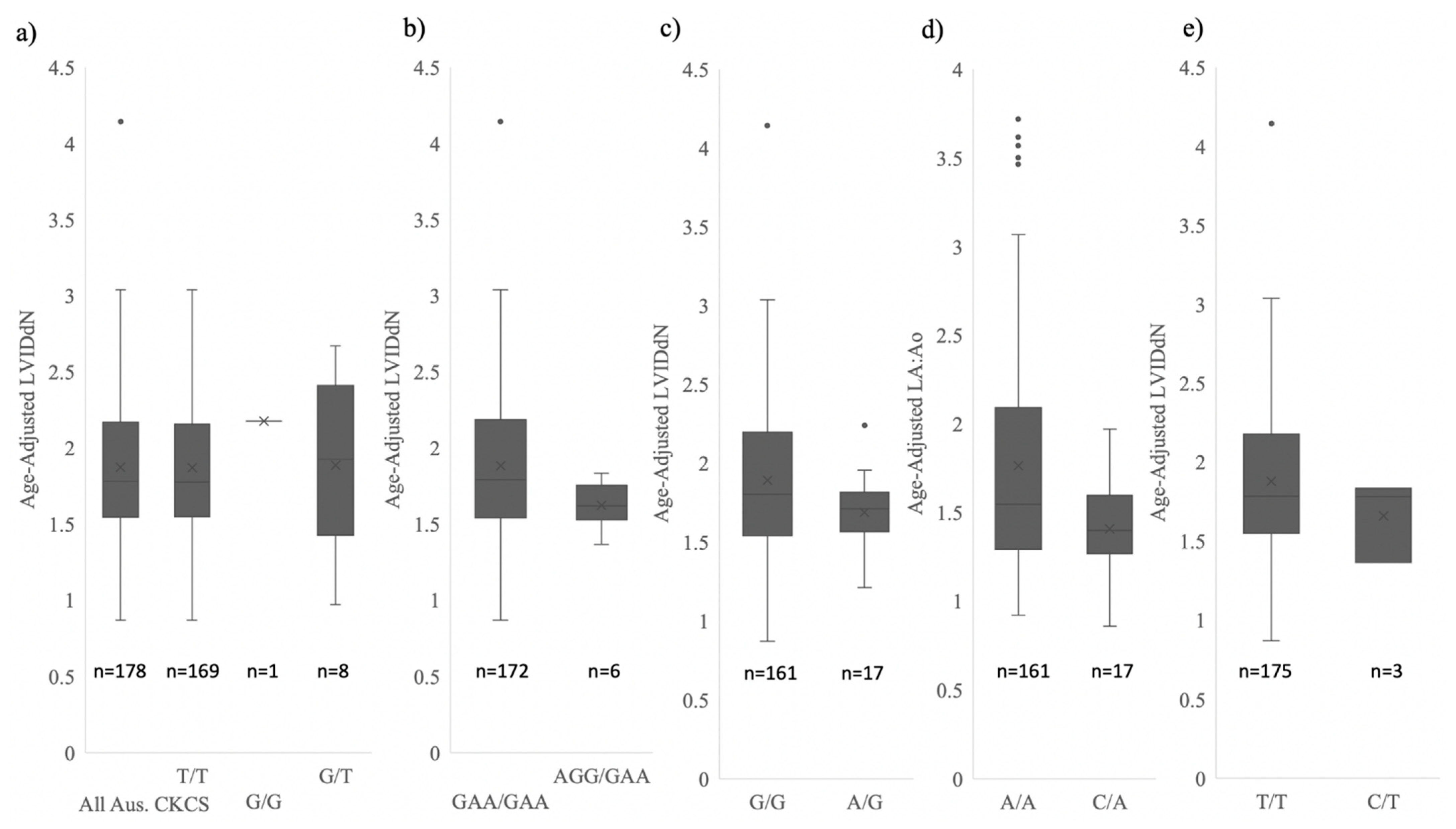
3.6. Analysis of Physiological Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fox, P. Pathology of myxomatous mitral valve disease in the dog. J. Vet. Cardiol. 2012, 14, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Borgarelli, M.; Savarino, P.; Crosara, S.; Santilli, R.; Chiavegato, D.; Poggi, M.; Bellino, C.; La Rosa, G.; Zanatta, R.; Haggstrom, J.; et al. Survival characteristics and prognostic variables of dogs with mitral regurgitation attributable to myxomatous valve disease. J. Vet. Intern. Med. 2008, 22, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M.; Elliott, C.; Loughran, K.; Kossar, A.; Castillero, E.; Levy, R.; Ferrari, G. Comparative pathology of human and canine myzomatous mitral valve degeneration: 5HT and TGF-beta mechanisms. Cardiovasc. Pathol. 2020, 46, 107196. [Google Scholar] [CrossRef]
- Keene, B.W.; Atkins, C.E.; Bonagura, J.D.; Fox, P.R.; Häggström, J.; Fuentes, V.L.; Oyama, M.A.; Rush, J.E.; Stepien, R.; Uechi, M. ACVIM consensus guidelines for the diagnosis and treatment of myxomatous mitral valve disease in dogs. J. Vet. Int. Med. 2019, 33, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Borgarelli, M.; Buchanan, J. Historical review, epidemiology and natural history of degenerative mitral valve disease. J. Vet. Cardiol. 2012, 14, 93–101. [Google Scholar] [CrossRef]
- Axelsson, E.; Ljungvall, I.; Bhoumik, P.; Conn, L.B.; Muren, E.; Ohlsson, Å.; Olsen, L.H.; Engdahl, K.; Hagman, R.; Hanson, J.; et al. The genetic consequences of dog breed formation-accumulation of deleterious genetic variation and fixation of mutations associated with myxomatous mitral valve disease in Cavalier King Charles Spaniels. PLoS Genet. 2021, 17, e.1009726. [Google Scholar] [CrossRef]
- Lewis, T.; Swift, S.; Woolliams, J.; Blott, S. Heritability of premature mitral valve disease in Cavalier King Charles Spaniels. Vet. J. 2011, 188, 73–76. [Google Scholar] [CrossRef]
- Summers, J.F.; O’Neill, D.G.; Church, D.B.; Thomson, P.C.; McGreevy, P.D.; Brodbelt, D.C. Prevalence of disorders recorded in Cavalier King Charles Spaniels attending primary-care veterinary practices in England. Canine Genet. Epidemiol. 2015, 2, 4. [Google Scholar] [CrossRef]
- French, A.T.; Ogden, R.; Eland, C.; Hemani, G.; Pong-Wong, R.; Corcoran, B.; Summers, K.M. Genome-wide analysis of mitral valve disease in Cavalier King Charles Spaniels. Vet. J. 2012, 193, 283–286. [Google Scholar] [CrossRef]
- Markby, G.R.; Macrae, V.E.; Corcoran, B.M.; Summers, K.M. Comparative transciptomic profiling of myxomatous mitral valve disease in the Cavalier King Charles Spaniel. BMC Vet. Res. 2020, 16, 350. [Google Scholar] [CrossRef]
- Bionda, A.; Cortellari, M.; Bagardi, M.; Frattini, S.; Negro, A.; Locatelli, C.; Brambilla, P.; Giuseppina, P.; Crepaldi, P. A genomic study of myxomatous mitral valve disease in Cavalier King Charles Spaniels. Animals 2020, 10, 1895. [Google Scholar] [CrossRef]
- Jung, S.; Bohan, A. Genome-wide sequencing and quantification of circulating microRNAs for dogs with congestive heart failure secondary to myxomatous mitral valve degeneration. Am. J. Vet. Res. 2018, 79, 163–169. [Google Scholar] [CrossRef]
- Swenson, L.; Häggström, J.; Kvart, C.; Juneja, R. Relationship between parental cardiac status in Cavalier King Charles Spaniels and prevalence and severity of chronic vascular disease in offspring. J. Am. Vet. Med. Assoc. 1996, 208, 2009–2012. [Google Scholar]
- Madsen, M.B.; Olsen, L.H.; Häggström, J.; Höglund, K.; Ljungvall, I.; Falk, T.; Wess, G.; Stephenson, H.; Dukes-McEwan, J.; Chetboul, V.; et al. Identification of 2 loci associated with development of myxomatous mitral valve disease in Cavalier King Charles Spaniels. J. Hered. 2011, 102, s62–s67. [Google Scholar] [CrossRef]
- Meurs, K.M.; Friedenberg, S.G.; Williams, B.; Keene, B.W.; Atkins, C.E.; Adin, D.; Aona, B.; DeFrancesco, T.; Tou, S.; Mackay, T. Evaluation of genes associated with human myxomatous mitral valve disease in dogs with familial myxomatous mitral valve degeneration. Vet. J. 2018, 232, 16–19. [Google Scholar] [CrossRef]
- Lu, C.-C.; Liu, M.-M.; Culshaw, G.; Clinton, M.; Argyle, D.J.; Corcoran, B.M. Gene network and canonical pathway analysis in canine myxymatous mitral valve disease: A microarray study. Vet. J. 2015, 204, 23–31. [Google Scholar] [CrossRef]
- Jagannathan, V.; Gemüller, C.; Leeb, T.; Aguirre, G.; Ané, C.; Bannasch, D.; Becker, D.; Davis, B.; Eckenstedt, K.; Faller, K.; et al. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef]
- Ancot, F.; Lemay, P.; Knowler, S.P.; Kennedy, K.; Griffiths, S.; Cherubini, G.B.; Sykes, J.; Mandigers, P.J.J.; Rouleau, G.A.; Rusbridge, C.; et al. A genome-wide association study identifies candidate loci associated to syringomyelia secondary to Chari-like malformation in Cavalier King Charles Spaniels. BMC Genet. 2018, 19, 16. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Microsoft Excel. Available online: https://office.microsoft.com/excel (accessed on 7 July 2022).
- Cunningham, F.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.; Armean, I.; Austine-Orimoloye, O.; Azov, A.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Boswood, A. Improving outcomes of myxomatous mitral valve disease in dogs. Practice 2018, 40, 12–15. [Google Scholar] [CrossRef]
- Maiellaro-Rafferty, K.; Wansapura, J.P.; Mendisaikhan, U.; Osinska, H.; James, J.F.; Taylor, M.D.; Robbins, J.; Kranias, E.G.; Towbin, J.A.; Purevjav, E. Altered regional cardiac wall mechanics are associated with differential cardiomyocyte calcium handling due to nebulette mutations in preclinical inherited dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2013, 60, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Tomasov, P.; Villard, E.; Faludi, R.; Melacini, P.; Lossie, J.; Lohmann, N.; Richard, P.; De Bortoli, M.; Angelini, A.; et al. Mutations in NEBL encoding the cardiac Z-disk protein nebulette are associated with various cardiomyopathies. Arch. Med. Sci. 2016, 12, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Purevjav, E.; Varela, J.; Morgado, M.; Kearney, D.L.; Li, H.; Taylor, M.D.; Arimura, T.; Moncman, C.K.; McKenna, W.; Murphy, R.T.; et al. Nebulette mutations are associated with dilated cardiomyopathy and endocardial fibroelastosis. J. Am. Coll. Cardiol. 2010, 56, 1493–1502. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Region | Variant | Consequence (Axelsson et al. [1] for Risk Variants) | Position (canFam3.1) | Reference Allele (canFam3) | Alternate Allele | Genotype | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3256 | 3261 | 3262 | 3333 | 3334 | 3336 | 80271 | 80294 | ||||||
| Nebulette | NEBL1 risk variant | Regulation | chr2:11 816 535 | A | G | A/G | G/G | G/G | A/G | A/G | G/G | A/G | A/G |
| Nebulette | New variant | Discovery | chr2:11 816 545 | G | A | A/G | G/G | G/G | A/G | A/G | G/G | A/G | A/G |
| Nebulette | NEBL1 predictive marker | Array marker | chr2:11 822 980 | A | G | A/G | G/G | G/G | A/G | A/G | G/G | A/G | A/G |
| Nebulette | NEBL2 risk variant | Regulation | chr2:11 823 576 | C | T | - | - | - | C/T | C/T | - | C/T | C/T |
| Nebulette | NEBL2 predictive marker | Array marker | chr2:11 832 538 | G | A | G/A | A/A | A/A | G/A | G/A | A/A | G/A | G/A |
| Nebulette | New variant | Discovery | chr2:11 979 669 | G | T | T/G | T/T | T/T | T/G | T/G | T/T | T/G | T/G |
| Nebulette | NEBL3 risk variant (and predictive marker) | Candidate mutation | chr2:11 979 724 | G | A | G/A | A/A | A/A | G/A | G/A | A/A | G/A | G/A |
| Hepatoma Derived Growth Factor-Like 1 | HDGFL1 risk variant | Candidate mutation | chr7:41 245 057 | A | G | G/A | G/A | A/A | G/G | G/G | G/A | G/G | G/G |
| Hepatoma Derived Growth Factor-Like 1 | HDGFL1 predictive marker | Array marker | chr7:41 248 384 | T | G | G/T | G/T | G/G | T/T | T/T | G/T | T/T | T/T |
| Gene | Variant | Position | Reference Allele Frequency CKCS a | Reference Allele | Alternative Allele | Representative Genotyping Array Location | CKCS Observed Mean MAF (N = 180 Array) |
|---|---|---|---|---|---|---|---|
| Nebulette | NEBL1 | chr2:11 816 535 | 0.00 | A | G | chr2:11 822 980 | 0.02 |
| Nebulette | NEBL2 | chr2:11 823 576 | 0.00 | C | T | chr2:11 832 538 | 0.02 |
| Nebulette | NEBL3 | chr2:11 979 724 | 0.00 | G | A | chr2:11 979 724 | 0.02 |
| Nebulette | NEBL4 | chr2:12 082 890 | 0.99 | T | C | chr2:12 085 928 | 0.05 |
| Nebulette | NEBL5 | chr2:12 165 498 | 0.94 | A | T | chr2:12 174 951 | 0.05 |
| Nebulette | NEBL6 | chr2:12 567 546 | 0.03 | A | G | chr2:12 567 760 | 0.01 |
| Latrophilin 2 | LPHN2 | chr6:65 609 405 | 0.17 | T | C | chr6:65 607 149 | 0.14 |
| Hepatoma Derived Growth Factor-Like 1 | HDGFL1 | chr7:41 245 057 | 0.00 | A | G | chr7:41 248 384 | 0.03 |
| Sorbin and SH3 Domain Containing 2 | SORBS2 | chr16:45 026 823 | 0.25 | C | T | chr16: 45 035 960 | 0.13 |
| Hydroxytryptamine Receptor 1F | HTR1F | chr31:273 549 | 0.15 | T | C | chr31:273 549 | 0.27 |
| Gene | Number of Markers | Mean Regional Minor Allele Frequency (±SD) | Accepted Status for Analysis |
|---|---|---|---|
| NEBL | 91 | 0.01 (±0.01) | Potential fixation |
| LPHN2 | 97 | 0.19 (±0.18) | Polymorphic |
| HDGFL1 | 153 | 0.03 (±0.05) | Potential fixation |
| SORBS2 | 126 | 0.14 (±0.18) | Polymorphic |
| HTR1F | 65 | 0.12 (±0.10) | Polymorphic |
| Variant | Forward Primer | Reverse Primer |
|---|---|---|
| NEBL1 | GGAAGCAGGCTCAGACTCTC | AACCTGACCAGTCCTTGGTG |
| NEBL2 | GCAGAAGGGCAACACTCTCT | TCTCTTTCTTTTGCCGCCCT |
| NEBL3 | AGCCCTCCTTCTGTGCTTTA | CTCCAAGGAGCCATCACATT |
| HDGFL1 | AGCACAGTCTCCCATCTCTC | CTCTCCAGGGGCTCTCTG |
| Variable | Total Cohort N = 178 | NEBL1-3 Homozygotes N = 172 | NEBL1-3 Heterozygotes N = 6 |
|---|---|---|---|
| Total Males | 80 (45%) | 78 (45%) | 2 (33%) |
| Total Females | 98 (55%) | 94 (55%) | 4 (67%) |
| Total ACVIM Stage A | 3 (2%) | 2 (1%) | 1 (17%) |
| Total ACVIM Stage B1 | 95 (53%) | 91 (53%) | 4 (67%) |
| Total ACVIM Stage B2 | 40 (22%) | 39 (23%) | 1 (17%) |
| Total ACVIM Stage C | 36 (20%) | 36 (21%) | 0 (0%) |
| Total ACVIM Stage D | 4 (2%) | 4 (2%) | 0 (0%) |
| Mean Age (±SD) | 10 ± 1.96 | 10 ± 1.97 | 10 ± 1.87 |
| Mean Age-Adjusted LA:Ao (±SD) | 1.73 ± 0.62 | 1.74 ± 0.63 | 1.37 ± 0.21 |
| Mean Age-Adjusted LVIDdN (±SD) | 1.87 ± 0.46 | 1.88 ± 0.46 | 1.62 ± 0.16 |
| Gene Variant | Location (CF3) | REF a | ALT b | Reference Allele Frequency (Wolves, N = 8) c | Reference Allele Frequency (Domestic dogs, N = 582) c | Reference Allele Frequency (CKCS, N = 4) c | Reference Allele Frequency (CKCS, N = 96) d |
|---|---|---|---|---|---|---|---|
| NEBL1 | chr2:11816535 | A | G | 0.81 | 0.56 | 0.13 | |
| NEBL2 | chr2:11823576 | C | T | 0.69 | 0.59 | 0.00 | |
| NEBL3 | chr2:11979724 | G | A | 1.00 | 0.85 | 0.13 | 0.01 |
| NEBL4 | chr2:12082890 | T | C | 0.06 | 0.28 | 1.00 | |
| NEBL5 | chr2:12165498 | A | T | 0.06 | 0.21 | 1.00 | |
| NEBL6 | chr2:12567546 | A | G | 0.75 | 0.84 | 0.00 | |
| LPHN2 | chr6:65609405 | T | C | 1.00 | 0.95 | 0.38 | |
| HDGFL1 | chr7:41245057 | A | G | 1.00 | 0.72 | 0.00 | 0.01 |
| SORBS2 | chr16:45026823 | C | T | 1.00 | 0.99 | 0.50 | |
| HTR1F | chr31:273549 | T | C | 1.00 | 0.96 | 0.50 |
| Variant (Representative Array Marker) | Mean Homozygous Cohort | Mean Heterozygous Cohort | F-Test F Value | F-Test Critical F value | T-Test T Statistic |
|---|---|---|---|---|---|
| Age-Adjusted LA:Ao | |||||
| NEBL1,2,3 | 1.74 ± 0.63 | 1.37 ± 0.21 | 9.38 ** | 4.39 ** | 3.82 ** |
| NEBL4 | 1.76 ± 0.64 | 1.41 ± 0.26 1 | 6.09 ** | 2.05 ** | 4.41 ** |
| NEBL5 | 1.76 ± 0.64 | 1.41 ± 0.26 1 | 6.09 ** | 2.05 ** | 4.41 ** |
| NEBL6 | 1.73 ± 0.63 | 1.53 ± 0.16 1 | 16.11 | 19.49 | 1.99 |
| HDGFL1 | 1.72 ± 0.62 | 1.96 ± 0.70 1 | 1.26 | 1.99 | 1.02 |
| Age-Adjusted LVIDdN | |||||
| NEBL1,2,3 | 1.88 ± 0.46 | 1.62 ± 0.16 1 | 8.64 ** | 4.39 * | 3.52 ** |
| NEBL4 | 1.89 ± 0.47 | 1.69 ± 0.23 1 | 4.06 ** | 2.05 ** | 3.02 ** |
| NEBL5 | 1.89 ± 0.47 | 1.69 ± 0.23 1 | 4.06 ** | 2.05 ** | 3.02 ** |
| NEBL6 | 1.87 ± 0.46 | 1.66 ± 0.26 1 | 3.22 | 19.49 | 1.41 |
| HDGFL1 | 1.87 ± 0.45 | 1.92 ± 0.55 1 | 1.49 | 1.99 | 0.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mead, S.E.; Beijerink, N.J.; O’Brien, M.; Wade, C.M. Genetic Variants at the Nebulette Locus Are Associated with Myxomatous Mitral Valve Disease Severity in Cavalier King Charles Spaniels. Genes 2022, 13, 2292. https://doi.org/10.3390/genes13122292
Mead SE, Beijerink NJ, O’Brien M, Wade CM. Genetic Variants at the Nebulette Locus Are Associated with Myxomatous Mitral Valve Disease Severity in Cavalier King Charles Spaniels. Genes. 2022; 13(12):2292. https://doi.org/10.3390/genes13122292
Chicago/Turabian StyleMead, Sophie E., Niek J. Beijerink, Mitchell O’Brien, and Claire M. Wade. 2022. "Genetic Variants at the Nebulette Locus Are Associated with Myxomatous Mitral Valve Disease Severity in Cavalier King Charles Spaniels" Genes 13, no. 12: 2292. https://doi.org/10.3390/genes13122292
APA StyleMead, S. E., Beijerink, N. J., O’Brien, M., & Wade, C. M. (2022). Genetic Variants at the Nebulette Locus Are Associated with Myxomatous Mitral Valve Disease Severity in Cavalier King Charles Spaniels. Genes, 13(12), 2292. https://doi.org/10.3390/genes13122292