


The Complete Chloroplast Genome of Hypoestes forskaolii (Vahl) R.Br: Insights into Comparative and Phylogenetic Analyses within the Tribe Justiceae

Abstract
1. Introduction
2. Materials and Methods
2.1. Library Construction, Sequencing and Assembly
2.2. Gene Annotation
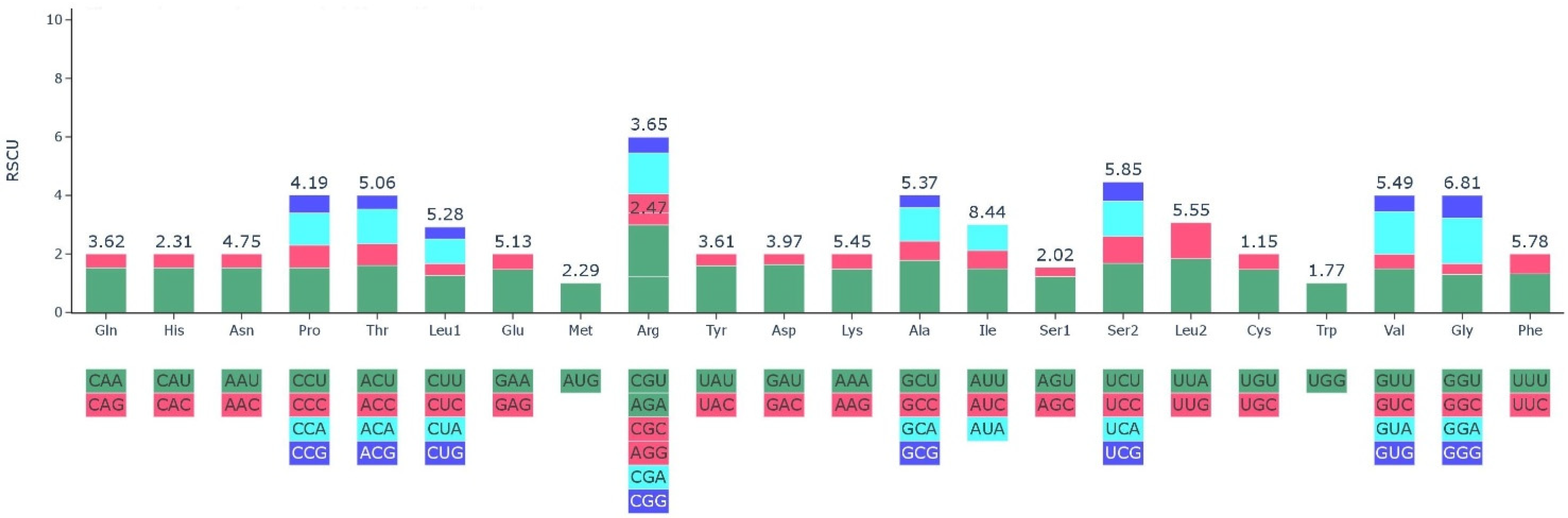
2.3. Codon Usage Analysis
2.4. Repeat Analysis
2.5. Genome Comparison
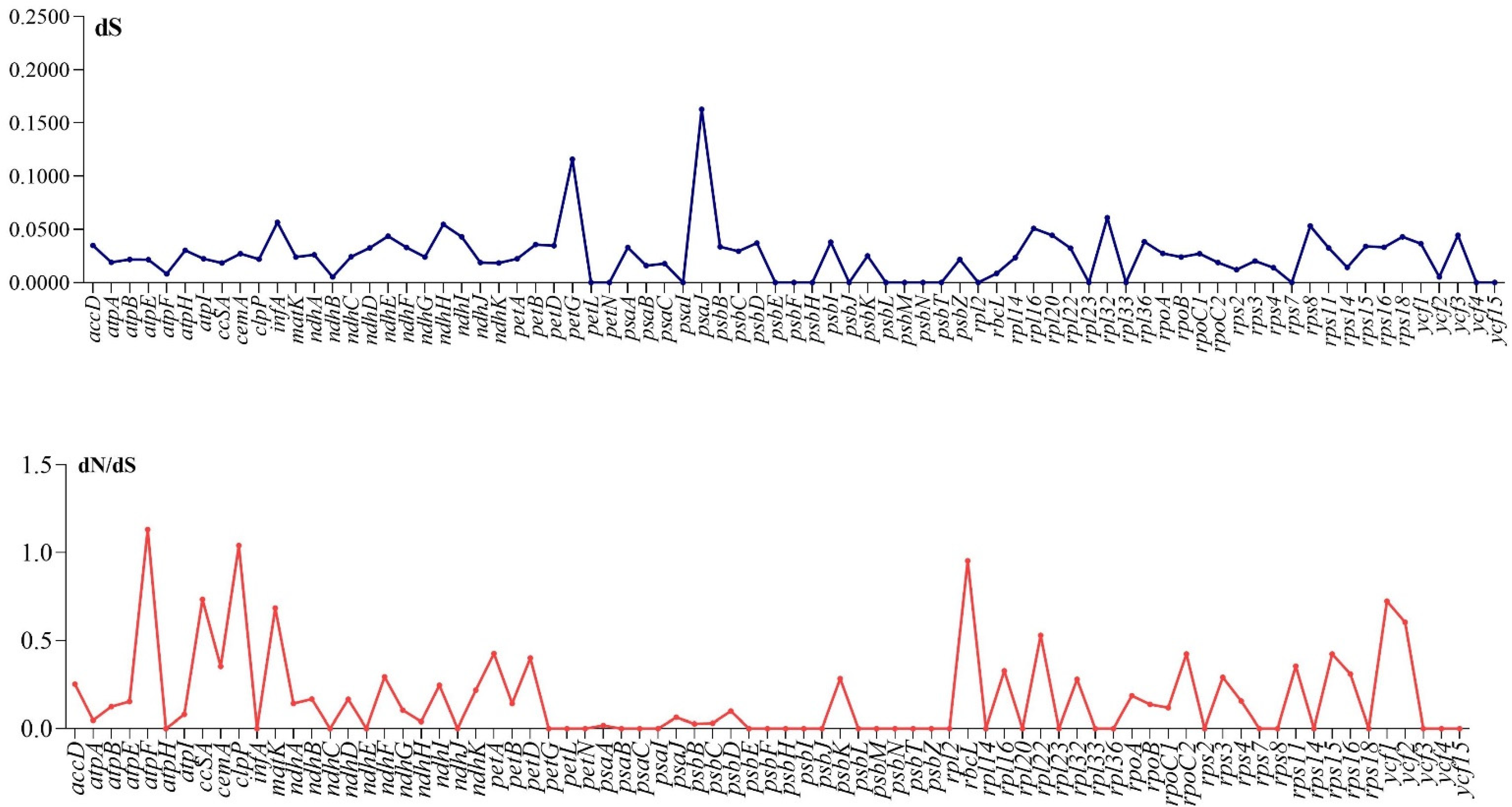
2.6. Characterization of Substitution Rate
2.7. Sequence Divergence
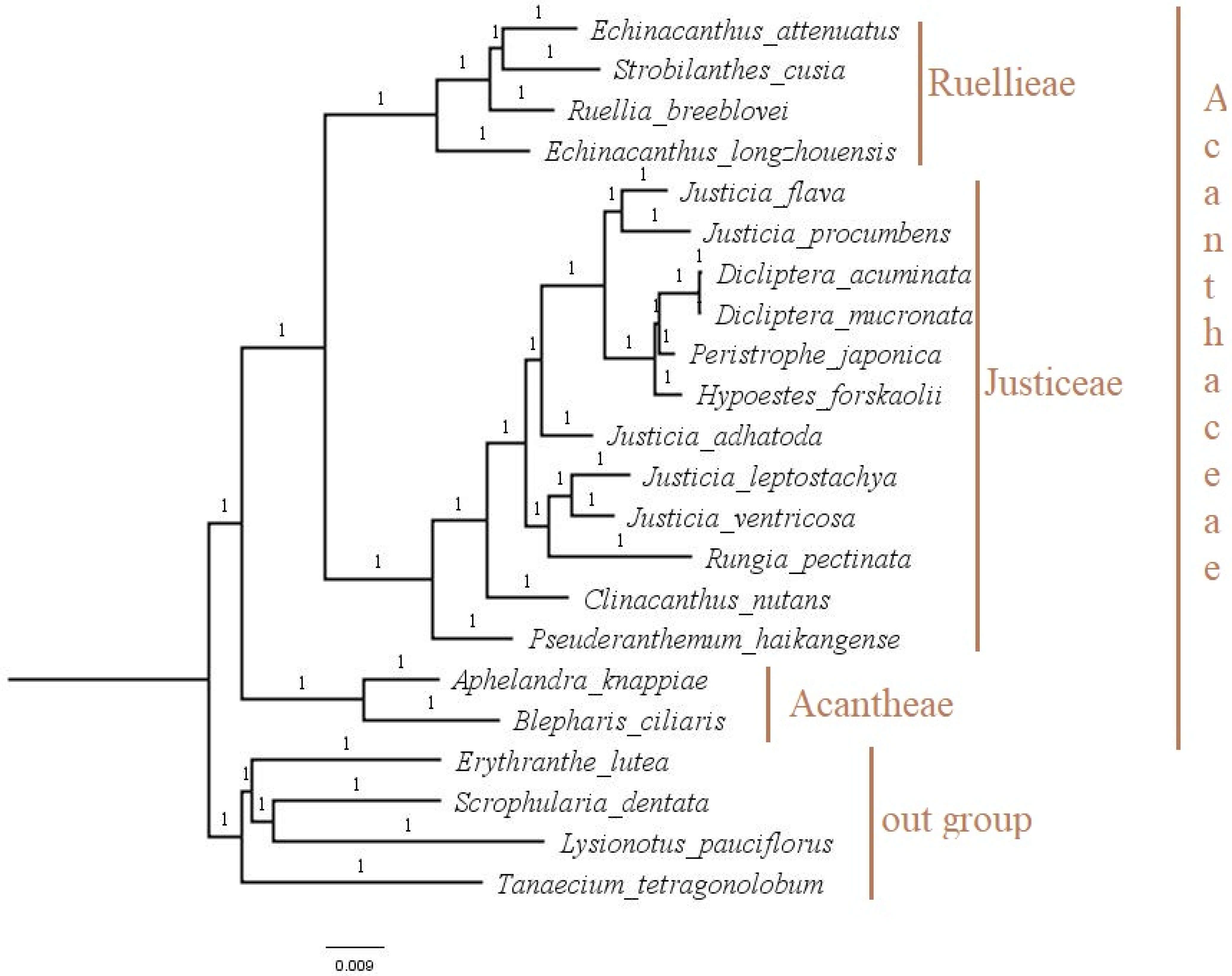
2.8. Phylogenetic Analysis
3. Results and Discussion
3.1. Characteristics of H. forskaolii Chloroplast Genome
3.2. Repeat Analyses
3.2.1. Long Repeats
3.2.2. Simple Sequence Repeats (SSRs)
3.3. Comparative Analysis of Justiceae Species Cp Genome
3.4. Divergence of Protein Coding Genes Sequence
3.5. Identification of Sequence Divergence
3.6. Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellis, B.W. Taylor’s Guide to Annuals: How to Select and Grow More Than 400 Annuals, Biennials, and Tender Perennials; Houghton Mifflin Co.: Boston, MA, USA, 1999; pp. 1–448. [Google Scholar]
- Almehdar, H.; Abdallah, H.M.; Osman, A.-M.M.; Abdel-Sattar, E.A. In Vitro cytotoxic screening of selected Saudi medicinal plants. J. Nat. Med. 2012, 66, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.C.; Ni, C.L.; Huang, Y.L.; Huang, R.L.; Chen, C.C. Furanolabdane diterpenes from Hypoestes purpurea. J. Nat. Prod. 2004, 67, 1947–1949. [Google Scholar] [CrossRef] [PubMed]
- Mothana, R.A.; Al-Musayeib, N.M.; Matheeussen, A.; Cos, P.; Maes, L. Assessment of the in vitro antiprotozoal and cytotoxic potential of 20 selected medicinal plants from the island of Soqotra. Molecules 2012, 17, 14349–14360. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, K.; Norris, F.G. Taxonomic studies in the Acanthaceae; The genus Hypoestes in southern Africa. S. Afr. J. Bot. 1985, 51, 133–144. [Google Scholar] [CrossRef]
- Padysakova, E.; Bartos, M.; Tropek, R.; Janecek, S. Generalization versus specialization in pollination systems: Vistors, thieves, and pollinators of hypoestes aristate (Acanthaceae). PLoS ONE 2013, 8, e59299. [Google Scholar] [CrossRef]
- Lindau, G. Acanthaceae. In Die natürlichen Pflanzenfamilien, IV(3b); Engler, A., Prantl, K., Eds.; Engelmann: Leipzig, Germany, 1895; pp. 274–354. [Google Scholar]
- Van Tieghem, P. Structure du pistil et de I’ovule du fruit et de la grane des Acathacees. Ann. Sci. Nat. Serie 9. Botanique. 1908, 7, 1–24. [Google Scholar]
- Bremekamp, C.E.B. Notes on some acanthaceous genera of controversial position. Acta Bot. Neerl. 1955, 4, 644–655. [Google Scholar] [CrossRef][Green Version]
- Bremekamp, C.E.B. Delimitation and subdivision of the Acanthaceae. Bull. Bot. Surv. India. 1965, 7, 21–30. [Google Scholar]
- Scotland, R.W.; Vollesen, K. Classification of Acanthaceae. Kew Bull. 2000, 55, 513–589. [Google Scholar] [CrossRef]
- McDade, L.A.; Daniel, T.F.; Masta, S.E.; Riley, K.M. Phylogenetic relationships within the tribe Justicieae (Acanthaceae): Evidence from molecular sequences, morphology, and cytology. Ann. Missouri Bot. Gard. 2000, 87, 435–458. [Google Scholar] [CrossRef]
- Kiel, C.A.; McDade, L.A.; Daniel, T.F.; Champluvier, D. Phylogenetic delimitation of Isoglossinae (Acanthaceae: Justicieae) and relationships among constituent genera. Taxon 2006, 55, 683–694. [Google Scholar] [CrossRef]
- Daniel, T.F.; McDade, L.A.; Manktelow, M.; Kiel, C.A. The “Tetramerium Lineage” (Acanthaceae: Acanthoideae: Justicieae): Delimitation and intra-lineage relationships based on cp and nrlTS sequence data. Syst. Bot. 2008, 33, 416–436. [Google Scholar] [CrossRef]
- Kiel, C.A.; McDade, L.A. The Mirandea clade (Acanthaceae, Justicieae, Tetramerium Lineage): Phylogenetic signal from molecular data and micromorphology makes sense of taxonomic confusion caused by remarkable diversity of floral form. Syst. Bot. 2014, 39, 950–964. [Google Scholar] [CrossRef]
- Côrtes, A.L.; Rapini, A.; Daniel, T.F. The Tetramerium lineage (Acanthaceae: Justicieae) does not support the Pleistocene Arc hypothesis for South American seasonally dry forests. Amer. J. Bot. 2015, 102, 992–1007. [Google Scholar] [CrossRef]
- Kiel, C.A.; Daniel, F.T.; Darbyshire, I.; McDade, L.A. Unraveling relationships in the morphologically diverse and taxonomically challenging “justicioid” lineage (Acanthaceae: Justicieae). Taxon 2017, 66, 645–674. [Google Scholar] [CrossRef]
- Neuhaus, H.; Emes, M. Nonphotosynthetic metabolism in plastids. Ann. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef]
- Grevich, J.J.; Daniell, H. Chloroplast Genetic Engineering: Recent Advances and Future Perspectives. Crit. Rev. Plant Sci. 2005, 24, 83–107. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Kai, F.M.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: Thetortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Chumley, T.W.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Implications of the plastid genome sequence of typha(Typhaceae, poales) for understanding genome evolution in poaceae. J. Mol. Evol. 2010, 70, 149–166. [Google Scholar] [CrossRef]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analysis. BMC Evol. Biol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. Geseq: Versatile and accurate annotation of organelle genomes. Nucl. Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0. Mol. Biol. Evol. 2015, 30, 2725–2729. [Google Scholar]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v6: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7:improvements in performance and usability. Mol. Biol.Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Fredrik, R.; Maxim, T.; Paul, V.M.; Daniel, L.A.; Aaron, D.; Sebastian, H.; Bret, L.; Liang, L.; Mar, A.S.; John, P.H. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference andModel Choice Across a Large Model Space. Systematic 2012, 61, 539–542. [Google Scholar]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shao, J.; Zhang, H.; Jiang, M.; Huang, L.; Zhang, Z.; Yang, D.; He, M.; Ronaghi, M.; Luo, X.; et al. Sequencing and analysis of Strobilanthes cusia (Nees) Kuntze chloroplast Genome revealed the rare simultaneous contraction and expansion of the inverted repeat region in Angiosperm. Front. Plant Sci. 2018, 9, 324. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, H.C.; Lin, I.P.; Chow, T.Y.; Chen, H.H.; Chen, W.H.; Cheng, C.H.; Lin, C.Y.; Liu, S.M.; Chang, C.C.; et al. The chloroplast genome of Phalaenopsis aphrodite (Orchidaceae): Comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications. Mol. Biol. Evol. 2006, 23, 279–291. [Google Scholar] [CrossRef]
- Samaila, S.Y.; Shah, M.M. The complete chloroplast genome of Lantana camara L. (Verbenaceae). Mitochondrial DNA part B 2020, 1, 919. [Google Scholar]
- Park, I.; Kim, W.J.; Yeo, S.-M.; Choi, G.; Kang, Y.-M.; Piao, R.; Moon, B.C. The complete chloroplast genome sequences of Fritillaria ussuriensis maxim. In addition, Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 2017, 282, 22. [Google Scholar]
- Amenu, S.G.; Wei, N.; Wu, L.; Oyebanji, O.; Hu., G.; Zhou, Y.; Wang, Q. Phylogenomic and comparative analyses of Coffeeae alliance (Rubiaceae): Deep insights into phylogenetic relationships and plastome evolution. BMC Plant Biol. 2022, 22, 88. [Google Scholar] [CrossRef]
- Campbell, W.H.; Gowri, G. Codon usage in higher plants, green algae, and cyanobacteria. Plant Physiol. 1990, 92, 1–11. [Google Scholar] [CrossRef]
- Liu, Q.; Dou, S.; Ji, Z.; Xue, Q. Synonymous codon usage and gene function are strongly related in Oryza sativa. Biosystems 2005, 80, 123–131. [Google Scholar] [CrossRef]
- Li, B.; Lin, F.R.; Huang, P.; Guo, W.Y.; Zheng, Y.Q. Complete chloroplast Genome sequence of Decaisnea insignis: Genome organization, Genomic resources and comparative analysis. Sci. Rep. 2017, 7, 10073. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; Shanker, A. Identification of simple sequence repeats in chloroplast genomes of Magnoliids through bioinformatics approach. Interdiscip. Sci. Comput. Life Sci. 2016, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, X.; Cui, Y.; Sun, W.; Li, Y.; Wang, Y. Molecular structure and phylogenetic analyses of complete chloroplast genomes of two Aristolochia medicinal species. Int. J. Mol. Sci. 2017, 18, 1839. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, Z.; Zhang, T.; Zhong, W.; Liu, C.; Yuan, Q.; Huang, L. The chloroplast genome sequence of Scutellaria baicalensis provides insight into intraspecific and interspecific chloroplast genome diversity in Scutellaria. Genes 2017, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Cui, Y.; Chen, X.; Li, Y.; Xu, Z.; Duan, B.; Li, Y.; Song, J.; Yao, H. Complete chloroplast genomes of Papaver rhoeas and Papaver orientale: Molecular structures, comparative analysis, and phylogenetic analysis. Molecules 2018, 23, 437. [Google Scholar] [CrossRef]
- Li, Y.G.; Xu, W.Q.; Zou, W.T.; Jiang, D.Y.; Liu, X.H. Complete chloroplast genome sequences of two endangered Phoebe(Lauraceae) species. Bot. Stud. 2017, 58, 37. [Google Scholar] [CrossRef]
- Greiner, S.; Wang, X.; Rauwolf, U.; Silber, M.V.; Mayer, K.; Meurer, J.; Haberer, G.; Herrmann, R.G. The complete nucleotide sequences of the five genetically distinct plastid genomes of Oenothera, subsection Oenothera: I. sequence evaluation and plastome evolution. Nucleic Acids Res. 2008, 36, 2366–2378. [Google Scholar] [CrossRef]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N. Chloroplast Genomic Resource of Paris for Species Discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef]
- Bryan, G.J.; McNicol, J.W.; Meyer, R.C.; Ramsay, G.; De Jong, W.S. Polymorphic simple sequence repeat markers in chloroplast genomes of Solanaceous plants. Theor. Appl. Genet. 1999, 99, 859–867. [Google Scholar] [CrossRef]
- Provan, J. Novel chloroplast microsatellites reveal cytoplasmic variation in Arabidopsis thaliana. Mol. Ecol. 2000, 9, 2183–2185. [Google Scholar] [CrossRef]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Jacinta, N.M.; Xiang, D.; Jia-Xin, Y.; Elijah, M.M.; Vincent, O.W.; Millicent, A.O.; Josphat, K.S.; Paulm, M.M.; Guang-Wan, H. Complete chloroplast genome of Chlorophytum comosum abd Chlorophytum gallabatense: Geome structures, comparative and phylogenetic analysis. Plants 2020. [Google Scholar]
- Dhafer, A.A.; Samaila, S.Y.; Enaj, J.A.; Abidina, A. Complete chloroplast genome sequence of Barleria prionitis, Comparative chloroplast genomics and phylogenetic relationships among Acanthoideae. BMC Genom. 2020, 21, 393. [Google Scholar]
- Philippe, H.; Delsuc, F.; Brinkmann, H.; Lartillot, N. Phylogenomics, Annual Review of Ecology. Evol. Sys. 2005, 36, 541–562. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boorem, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nupharadvena and Ranunculus macranthus. BMC Genom. 2007, 8, 174–201. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.; Bai, G.; Li, Z.; Kanwal, Z.; Zhao, G. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia Species. Front. Plant Sci. 2016, 7, 1512. [Google Scholar] [CrossRef]
- Rousseau-Gueutin, M.; Bellot, S.; Martin, G.E.; Boutte, J.; Chelaifa, H.; Lima, O.; Michon-Coudouel, S.; Naquin, D.; Salmon, A.; Ainouche, K.; et al. The chloroplast genome of the hexaploid Spartina maritima (Poaceae, Chloridoideae): Comparative analyses and molecular dating. Mol. Phylogenet. Evol. 2015, 93, 516. [Google Scholar] [CrossRef]
- Xu, J.-H.; Liu, Q.; Hu, W.; Want, T.; Xue, Q.; Messing, J. Dynamics of chloroplast genome in green plants. Genomics 2015, 106, 221–231. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Ratnasingham, S.; de Waard, J.R. Barcoding animal life: Cytochrome C oxidase subunit 1 divergences among closely related species. Proc. R. Soc. B Biol. Sci. 2003, 270, S96–S99. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Stoeckle, M.Y.; Zemlak, T.S.; Francis, C.M.; Charles, G. Identification of birds through DNA barcodes. PLoS Biol. 2004, 2, e312. [Google Scholar] [CrossRef]
- Borsch, T.; Quandt, D. Mutational dynamics and phylogenetic utility of noncoding chloroplast DNA. Plant Syst. Evol. 2009, 282, 169–199. [Google Scholar]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Kim, T.S.; Park, Y.J. Rice chloroplast genome variation architecture and phylogenetic dissection in diverse Oryza species assessed by whole-genome resequencing. Rice 2016, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Liu, H.; Xu, C.; Zuo, Y.J.; Chen, Z.J.; Zhou, S.L. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.P.; Bi, Y.; Yang, F.P.; Zhang, M.F.; Chen, X.Q.; Xue, J.; Zhang, X.H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef]
- Darbyshire, I.; Vollesen, K. The transfer of the genus Peristrophe to Dicliptera (Acanthaceae), with a new species described from eastern Africa. Kew Bull. 2007, 62, 119–128. [Google Scholar]
- Balkwill, K.F. Taxonomic studies in the tribe Justicieae of the family Acanthaceae. [Unpublished]. Ph.D. Thesis, University of Natal, Pietermaritzburg, South Africa, 1985. [Google Scholar]
- Balkwill, K.; Getiffe-Norris, F.; Balkwill, M.J. Taxonomic studies in the Acanthaceae: Dicliptera in southern Africa. Kew Bull. 1996, 51, 1–61. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | T(U) (%) | C (%) | A (%) | G (%) | Total (bp) | |
|---|---|---|---|---|---|---|
| cp genome | 31.2 | 19.4 | 30.8 | 18.7 | 151,142 | |
| LSC | 32.4 | 18.5 | 31.16 | 17.5 | 83,176 | |
| SSC | 34.0 | 16.8 | 34.0 | 15.2 | 17,012 | |
| IRA | 28.4 | 22.5 | 28.3 | 20.9 | 25,477 | |
| IRB | 28.2 | 20.8 | 28.4 | 22.5 | 25,477 | |
| 1st Positiom | 31.2 | 19.4 | 30.1 | 19.0 | 50,381 | |
| 2nd Position | 31.0 | 19.4 | 31.3 | 18.3 | 50,381 | |
| 3rd Position | 31.0 | 19.2 | 30.9 | 18.6 | 50,381 |
| Category | Grpup of Genes | Name of Genes |
|---|---|---|
| RNA genes | ribosomal RNA genes (rRNA) | rrn5, rrn4.5, rrn16, rrn23 |
| Transfer RNA genes (tRNA) | trnL-UAG., trnK-UUU +, trnS-GCU, trnQ-UUG, trnV-UAC +, trnR UCU, trnD-GUC, trnE-UUC, trnC-GCA, trnM-CAU, trnY-GUA, trnT-GGU, trnG-GCC, trnfM-CAU, trnS-UGA, trnT-UGU, trnL-UAA +, trnF-GAA, trnW-CCA, trnP-UGG, trnP-GGG, trnL-CAA a, trnV-GAC a, trnI-GAU +,a, trnA-UGC +,a, trnR-ACG a, trnN-GUU a, trnH-GUG, trnG-UCC, trnS-GGA | |
| Ribosomal proteins | Small subunit of ribosome | rps19, rps8, rps14, rps7 a, rps2, rps11, rps2, rps14, rps18, rps16 +, rps15, rps12 a |
| Transcription | Large subunit of ribosome | rpl33, rpl16, rpl20, rpl32, rpl23 a, rpl22, rpl14, rpl36, rpl2 +,a |
| DNA dependent RNA polymerase | rpoC2, rpoB rpoA, rpoC1 +, | |
| Protein genes Other genes | Photosystem I | psaI, psaB, psaj, psaA, psaC, ycf3 ++ |
| Photosystem II | psbZ, psbK, psbF, psbD, psbL, psbN, psbH, psbT, psbJ, psbB, psbE, psbM, psbC, psbI, psbA | |
| Subunit of cytochrome | petL, petB, petN, petG, petA, petD | |
| Subunit of synthase | atpH, atpF, atpE, atpB +, atpA, atpI | |
| Chloroplast envelope membrabe protien | cemA | |
| NADH dehydrogenase | ndhK, ndhH, ndhI, ndhD, ndhB +,a ndhG, ndhF, ndhE, ndhC, ndhJ, ndhA+ | |
| Large subunit of rubisco | rbcL | |
| Subunit acetyl-coA carboxylase | accD | |
| ATP dependent protease subunit P | clpP ++ | |
| Maturase | matK | |
| C-type cytochrome systhesis | ccsA | |
| Translational initiation factor | infA | |
| Component of TIC complex | ycf1 a | |
| Hypothetical proteins | ycf2 a,ycf4, ycf15 a |
| Gene | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|
| trnK-UUU | LSC | 36 | 2453 | 36 | ||
| rps16 | LSC | 224 | 912 | 35 | ||
| trnS-CGA | LSC | 31 | 661 | 59 | ||
| atpF | LSC | 470 | 649 | 140 | ||
| rpoC1 | LSC | 1635 | 784 | 433 | ||
| ycf3 | LSC | 152 | 710 | 228 | 683 | 127 |
| trnL-UAA | LSC | 36 | 513 | 49 | ||
| trnV-UAC | LSC | 36 | 584 | 37 | ||
| clpP | LSC | 226 | 631 | 299 | 749 | 69 |
| petB | LSC | 5 | 687 | 653 | ||
| rpl2 | IR | 435 | 658 | 397 | ||
| ndhB | IR | 755 | 679 | 776 | ||
| rps12 | IR | 25 | 542 | 231 | ||
| trnI-GAU | IR | 41 | 939 | 34 | ||
| trnA-UGC | IR | 37 | 820 | 34 | ||
| ndhA | SSC | 538 | 989 | 552 |
| Codon | Amino Acid | RSCU | tRNA | Codon | Amino Acid | RSCU | tRNA |
|---|---|---|---|---|---|---|---|
| UUC | Phe | 0.67 | trnF-GAA | UAU | Tyr | 1.61 | trnY-GUA |
| UUU | Phe | 1.33 | UAC | Tyr | 0.39 | ||
| CUG | Leu | 0.42 | CAG | Gln | 0.47 | ||
| UUA | Leu | 1.84 | trnL-UAA | UAA | Stop | 1.55 | |
| CUA | Leu | 0.83 | CAA | Gln | 1.53 | trnQ-UUG | |
| CUU | Leu | 1.27 | trnL-UAG | CAU | His | 1.54 | trnH-GUG |
| CUC | Leu | 0.4 | CAC | His | 0.46 | ||
| UUG | Leu | 1.24 | trnL-CAA | UAG | Stop | 0.93 | |
| AUG | Met | 1 | trnM-CAU | AAG | Lys | 0.51 | |
| AUU | Ile | 1.5 | trnI-GAU | AAU | Asn | 1.54 | trnN-GUU |
| GUC | Val | 0.49 | GAC | Asp | 0.36 | ||
| AUA | Ile | 0.89 | trnI-CAU | AAA | Lys | 1.49 | trnK-UUU |
| AUC | Ile | 0.61 | AAC | Asn | 0.46 | ||
| GUU | Val | 1.5 | trnV-GAC | GAU | Asp | 1.64 | trnD-GUC |
| UCU | Ser | 1.68 | trnS-GGA | UGU | Cys | 1.46 | trnC-GCA |
| GUA | Val | 1.45 | GAA | Glu | 1.47 | trnE-UUC | |
| GUG | Val | 0.56 | trnV-UAC | GAG | Glu | 0.53 | |
| UCA | Ser | 1.21 | UGA | Stop | 0.52 | ||
| UCC | Ser | 0.92 | UGC | Cys | 0.54 | ||
| CCA | Pro | 1.1 | CGA | Arg | 1.41 | ||
| UCG | Ser | 0.67 | trnS-UGA | UGG | Trp | 1 | trnW-CCA |
| CCU | Pro | 1.51 | trnP-UGG | CGU | Arg | 1.2 | trnR-ACG |
| CCC | Pro | 0.78 | CGC | Arg | 0.41 | trnR-UCU | |
| GCA | Ala | 1.14 | GGA | Gly | 1.58 | ||
| CCG | Pro | 0.61 | CGG | Arg | 0.55 | ||
| ACU | Thr | 1.61 | AGA | Arg | 1.78 | ||
| ACC | Thr | 0.73 | AGG | Arg | 0.65 | ||
| GCC | Ala | 0.67 | GGC | Gly | 0.37 | ||
| ACG | Thr | 0.47 | trnT-UGU | AGC | Ser | 0.3 | |
| GCU | Ala | 1.75 | trnA-UGC | GGU | Gly | 1.27 | |
| ACA | Thr | 1.19 | trnT-GGU | AGU | Ser | 1.22 | trnS-GCU |
| GCG | Ala | 0.44 | GGG | Gly | 0.78 | trnG-UCC |
| cpSSR ID | Repeat Motif | Length (bp) | No. of Repeats | SSR Start Position |
|---|---|---|---|---|
| 1 | (A) 8 | 8 | 23 | 4087; 8109; 9324; 14,945; 15,707; 18,082; 22,395; 41,631; 64,140; 80,149; 95,787; 110,230; 111,185; 112,039; 112,263; 114,062; 114,556; 118,479; 125,329; 136,542; 151,007; 151,074 |
| 2 | (A) 9 | 9 | 9 | 7444; 11,725; 26,746; 43,488; 43,722; 66,819; 73,976; 88,658; 151,041 |
| 3 | (A) 10 | 10 | 5 | 7650; 12,601; 14,734; 27,880; 132,799 |
| 4 | (A) 12 | 12 | 1 | 15,729 |
| 5 | (A) 13 | 13 | 1 | 7927 |
| 6 | (A) 16 | 16 | 1 | 127,320 |
| 7 | (A) 17 | 17 | 1 | 13,063 |
| 8 | (C) 11 | 11 | 1 | 4477 |
| 9 | (C) 12 | 12 | 1 | 131,580 |
| 10 | (G) 8 | 8 | 2 | 4422; 57,979 |
| 11 | (G) 9 | 9 | 1 | 5964 |
| 12 | (G) 10 | 10 | 1 | 116,259 |
| 13 | (G) 11 | 11 | 1 | 74,737 |
| 14 | (G) 12 | 12 | 1 | 102,700 |
| 15 | (G) 16 | 16 | 1 | 66,828 |
| 16 | (T) 8 | 8 | 27 | 7431; 8012; 25,690; 30,519; 32,669; 35,551; 42,497; 45,300; 50,150; 59,760; 72,331; 74,672; 75,843; 81,161; 82,593; 82,893; 83,210; 83,277; 97,742; 109,264; 111,758; 112,374; 113,566; 123,714; 123,762; 123,859; 138,497 |
| 17 | (T) 9 | 9 | 14 | 16,042; 17,941; 28,559; 31,561; 42,560; 65,592; 77,186; 83,242; 109,326; 115,581; 123,423; 123,827; 123,846; 145,625 |
| 18 | (T) 10 | 10 | 9 | 11,804; 12,514; 30,933; 53,860; 68,645; 70,546; 101,483; 123,976; 125,541 |
| 19 | (T) 11 | 11 | 3 | 59,201;123,498; 123,792; |
| 20 | (T) 12 | 12 | 2 | 54,320; 124,361 |
| 21 | (T) 13 | 13 | 1 | 9522 |
| 22 | (T) 16 | 16 | 1 | 106,956 |
| 23 | (AT) 5 | 5 | 2 | 20,342; 46,477 |
| 24 | AT (7) | 7 | 2 | 7285; 13,415 |
| 25 | AT (9) | 9 | 1 | 75,422 |
| 26 | (TA) 5 | 5 | 3 | 19,311; 45,521; 45,967 |
| 27 | TA (6) | 6 | 3 | 30,631; 45,576; 82,952 |
| 28 | (ATA) 4 | 4 | 2 | 64,698; 149,097 |
| 29 | (TTC) 4 | 4 | 1 | 34,430 |
| 30 | (AAT) 4 | 4 | 1 | 63,219 |
| 31 | (TTA) 4 | 4 | 1 | 81,350 |
| 32 | (TAT) 4 | 4 | 1 | 85,183 |
| 33 | (TGA) 4 | 4 | 1 | 90,439 |
| 34 | (TCT) 5 | 5 | 1 | 124,766 |
| 35 | (ATC) 4 | 4 | 1 | 143,840 |
| 36 | (ATAA) 3 | 3 | 1 | 45,662 |
| 37 | (TAAA) 3 | 3 | 1 | 66,210 |
| 38 | (AAAC) 3 | 3 | 1 | 67,218 |
| 39 | (AAAT) 3 | 3 | 1 | 74,480 |
| 40 | (AATA) 3 | 3 | 1 | 113,175 |
| 41 | (AATC) 3 | 3 | 1 | 118,310 |
| 42 | (AATT) 3 | 3 | 1 | 122,706 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yaradua, S.S.; Yessoufou, K. The Complete Chloroplast Genome of Hypoestes forskaolii (Vahl) R.Br: Insights into Comparative and Phylogenetic Analyses within the Tribe Justiceae. Genes 2022, 13, 2259. https://doi.org/10.3390/genes13122259
Yaradua SS, Yessoufou K. The Complete Chloroplast Genome of Hypoestes forskaolii (Vahl) R.Br: Insights into Comparative and Phylogenetic Analyses within the Tribe Justiceae. Genes. 2022; 13(12):2259. https://doi.org/10.3390/genes13122259
Chicago/Turabian StyleYaradua, Samaila Samaila, and Kowiyou Yessoufou. 2022. "The Complete Chloroplast Genome of Hypoestes forskaolii (Vahl) R.Br: Insights into Comparative and Phylogenetic Analyses within the Tribe Justiceae" Genes 13, no. 12: 2259. https://doi.org/10.3390/genes13122259
APA StyleYaradua, S. S., & Yessoufou, K. (2022). The Complete Chloroplast Genome of Hypoestes forskaolii (Vahl) R.Br: Insights into Comparative and Phylogenetic Analyses within the Tribe Justiceae. Genes, 13(12), 2259. https://doi.org/10.3390/genes13122259