
A Multigraph-Based Representation of Hi-C Data



Abstract
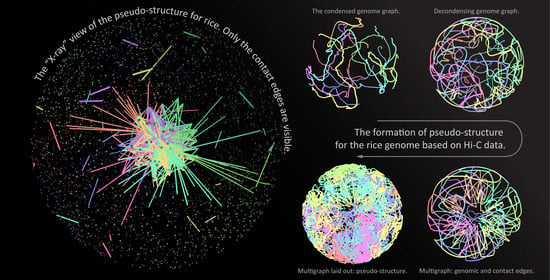
{kind=link}
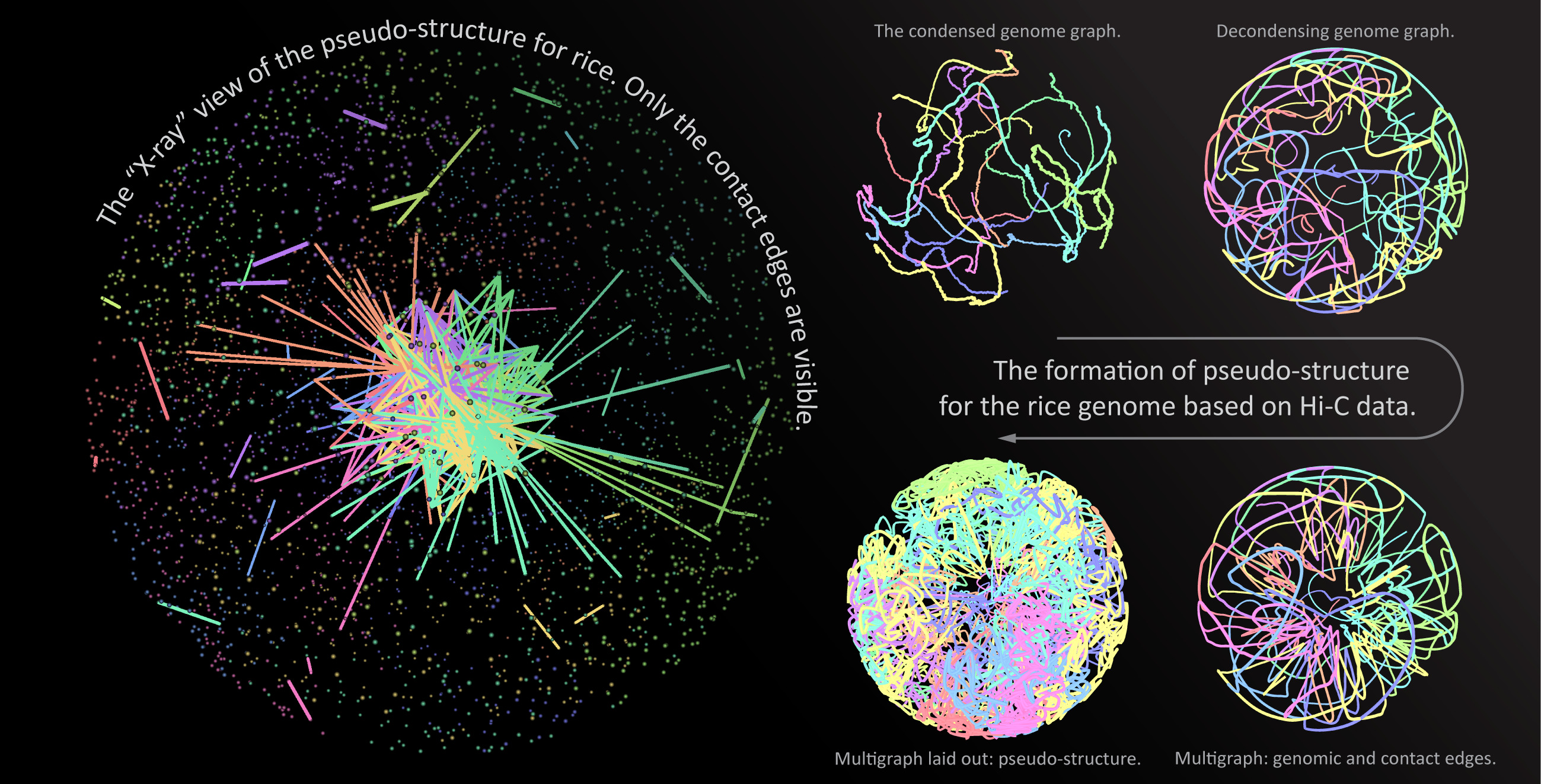
{kind=link}
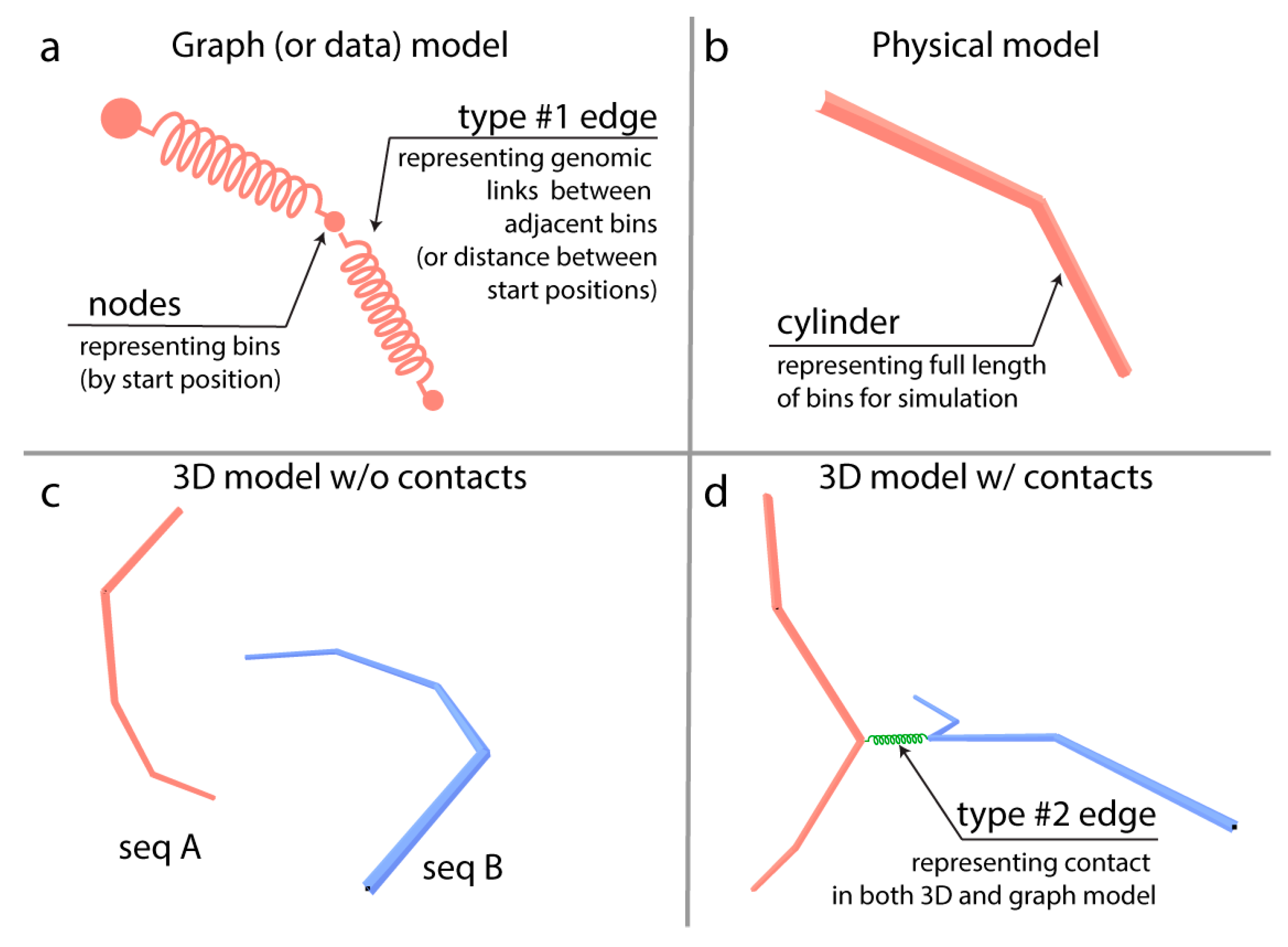
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Raw Data Analyses
2.2. Contact Statistics
2.3. Multigraph Construction
2.4. Calculation of the 2D/3D Layout
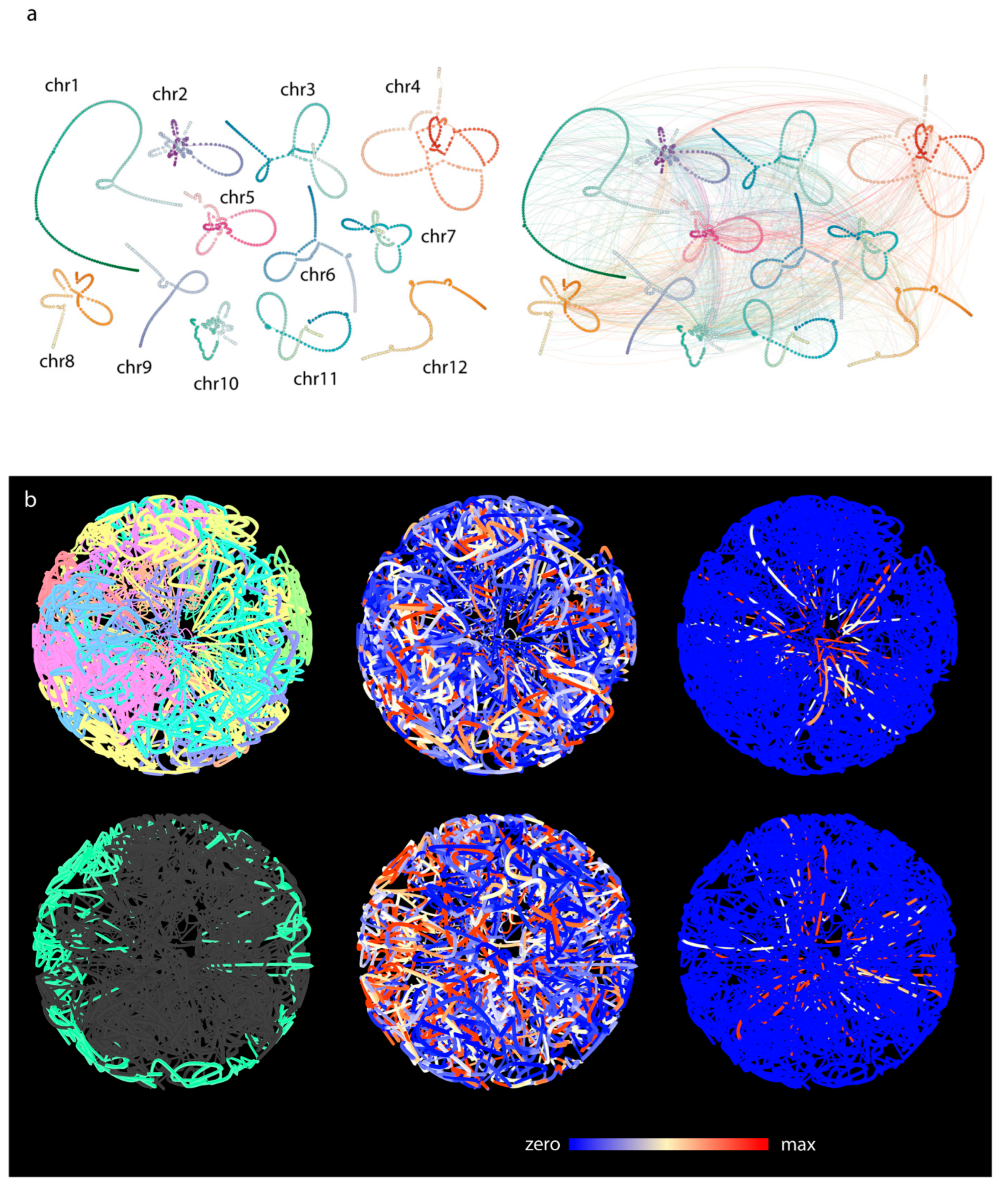
2.5. Chromosome Territories
2.6. Cytological Analysis
3. Results
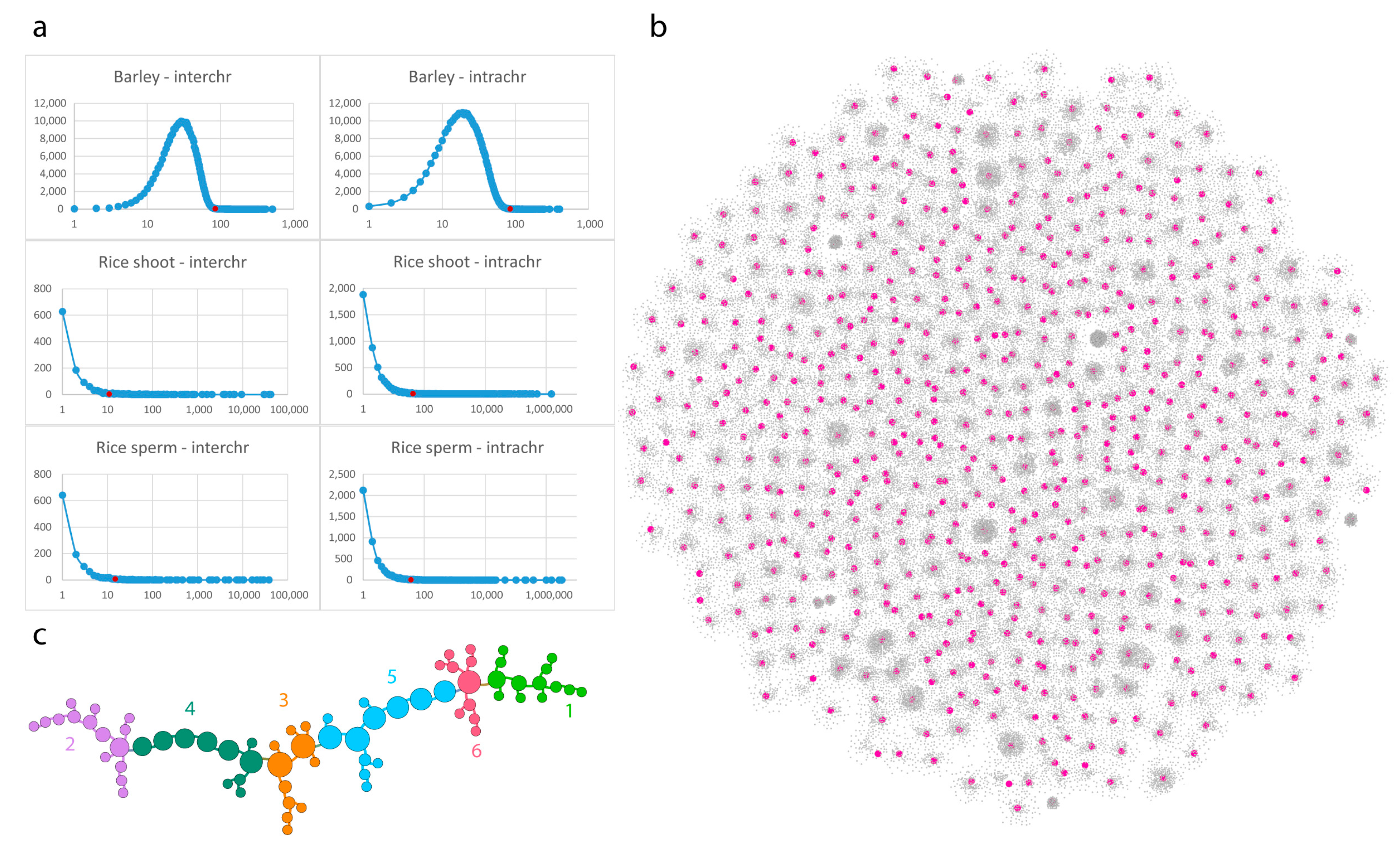
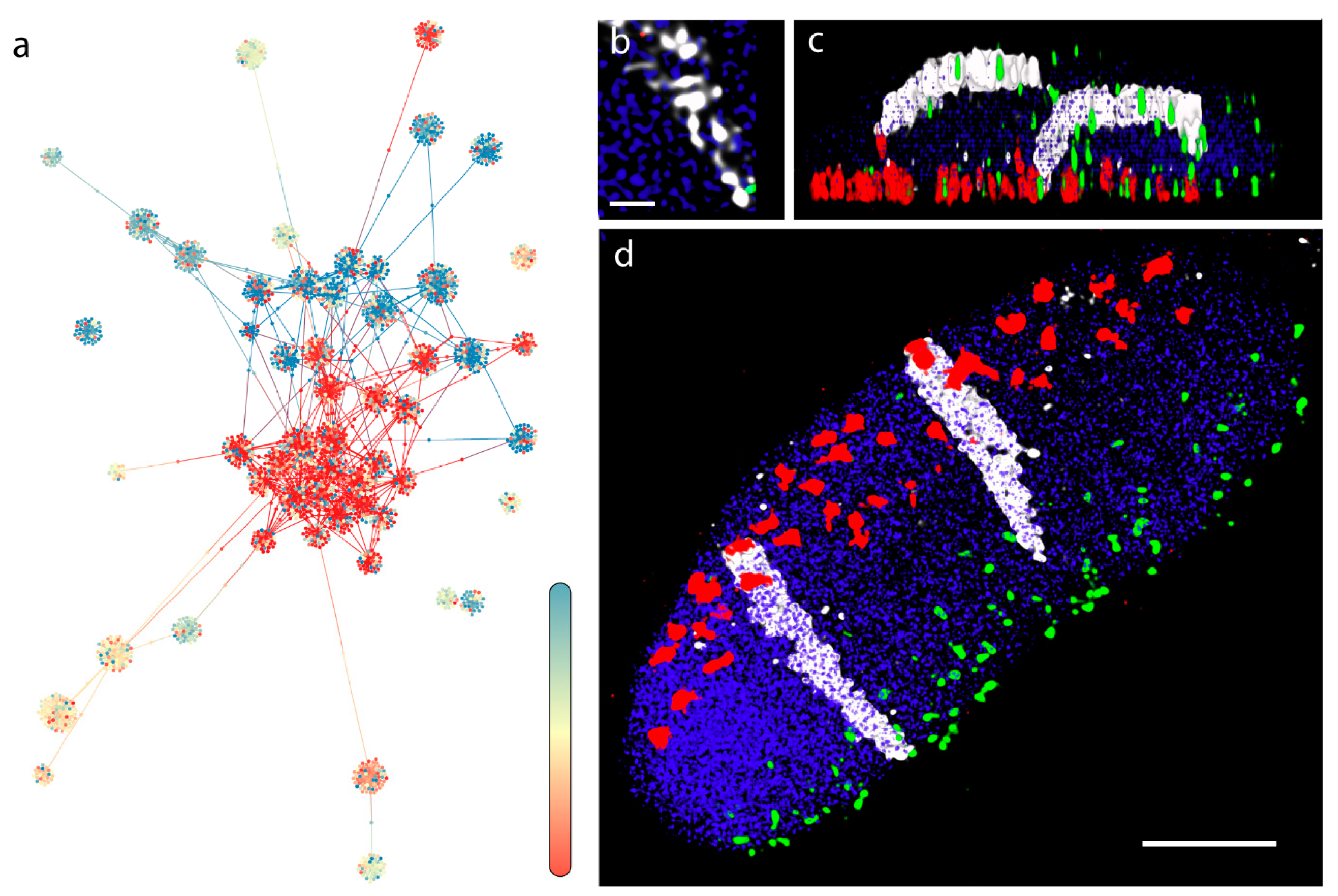
3.1. Barley (Multicellular, Bulk Samples)
3.2. Three-Dimensional Layout of the Multigraph Representation of the Genome Reflects Chromosome Territories and a Polarized Configuration
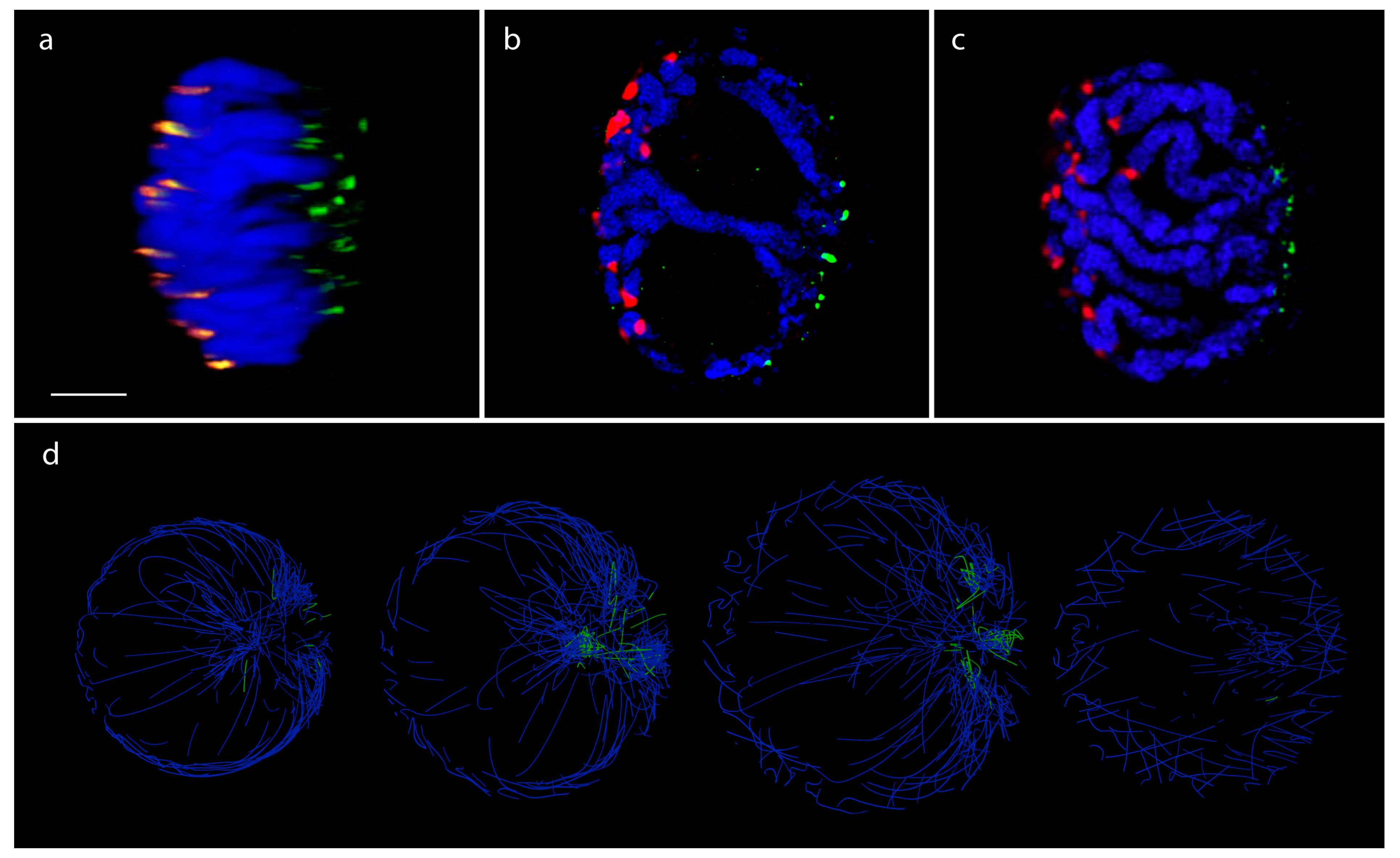
3.3. Rice (Single Cell Experiments)
3.4. Pure Contact Graphs of Rice
3.5. Multigraph Representation of Single Cell Hi-C Data of Rice
3.6. Comparative Analysis of Barley and Rice Hi-C Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heslop-Harrison, J.S.P.; Schwarzacher, T. Organisation of the Plant Genome in Chromosomes. Plant J. 2011, 66, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Gibcus, J.H.; Samejima, K.; Goloborodko, A.; Samejima, I.; Naumova, N.; Nuebler, J.; Kanemaki, M.T.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. A Pathway for Mitotic Chromosome Formation. Science 2018, 359, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.R.; Hudson, D.F.; Cisneros-Soberanis, F.; Earnshaw, W.C. Mitotic Chromosomes. Semin. Cell Dev. Biol. 2021, 117, 7–29. [Google Scholar] [CrossRef] [PubMed]
- Beseda, T.; Cápal, P.; Kubalová, I.; Schubert, V.; Doležel, J.; Šimková, H. Mitotic Chromosome Organization: General Rules Meet Species-Specific Variability. Comput. Struct. Biotechnol. J. 2020, 18, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C Reveals That CTCF Underlies Evolution of Chromosomal Domain Architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef]
- Liu, C.; Wang, C.; Wang, G.; Becker, C.; Zaidem, M.; Weigel, D. Genome-Wide Analysis of Chromatin Packing in Arabidopsis Thaliana at Single-Gene Resolution. Genome Res. 2016, 26, 1057–1068. [Google Scholar] [CrossRef]
- Grob, S.; Schmid, M.W.; Grossniklaus, U. Hi-C Analysis in Arabidopsis Identifies the KNOT, a Structure with Similarities to the Flamenco Locus of Drosophila. Mol. Cell 2014, 55, 678–693. [Google Scholar] [CrossRef]
- Rosa, S.; Shaw, P. Insights into Chromatin Structure and Dynamics in Plants. Biology 2013, 2, 1378–1410. [Google Scholar] [CrossRef]
- Dong, P.; Tu, X.; Chu, P.Y.; Lü, P.; Zhu, N.; Grierson, D.; Du, B.; Li, P.; Zhong, S. 3D Chromatin Architecture of Large Plant Genomes Determined by Local A/B Compartments. Mol. Plant 2017, 10, 1497–1509. [Google Scholar] [CrossRef]
- Tsompana, M.; Buck, M.J. Chromatin Accessibility: A Window into the Genome. Epigenetics Chromatin 2014, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of Nucleosome Dynamics by Histone Modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Over, R.S.; Michaels, S.D. Open and Closed: The Roles of Linker Histones in Plants and Animals. Mol. Plant 2014, 7, 481–491. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Sazer, S.; Schiessel, H. The Biology and Polymer Physics Underlying Large-Scale Chromosome Organization. Traffic 2018, 19, 87–104. [Google Scholar] [CrossRef]
- Carstens, S.; Nilges, M.; Habeck, M. Inferential Structure Determination of Chromosomes from Single-Cell Hi-C Data. PLoS Comput. Biol. 2016, 12, e1005292. [Google Scholar] [CrossRef]
- Das, P.; Shen, T.; McCord, R.P. Inferring Chromosome Radial Organization from Hi-C Data. BMC Bioinform. 2020, 21, 511. [Google Scholar] [CrossRef]
- Di Stefano, M.; Nützmann, H.W. Modeling the 3D Genome of Plants. Nucleus 2021, 12, 65–81. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of Entangled Linear Polymer Melts: A Molecular-Dynamics Simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The INO80 Remodeller in Transcription, Replication and Repair. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160290. [Google Scholar] [CrossRef]
- Ehrenhofer-Murray, A.E. Chromatin Dynamics at DNA Replication, Transcription and Repair. Eur. J. Biochem. 2004, 271, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Fisher, L.M. DNA Unwinding in Transcription and Recombination. Nature 1982, 299, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Monem, M.; Dürwald, H.; Hoffmann-Berling, H. Enzymic Unwinding of DNA: 2. Chain Separation by an ATP-Dependent DNA Unwinding Enzyme. Eur. J. Biochem. 1976, 65, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Hooke, R. Lectiones Cutlerianae, or A Collection of Lectures: Physical, Mechanical, Geographical, & Astronomical; Printed for John Martyn: London, UK, 1679. [Google Scholar]
- Schreiber, M.; Mascher, M.; Wright, J.; Padmarasu, S.; Himmelbach, A.; Heavens, D.; Milne, L.; Clavijo, B.J.; Stein, N.; Waugh, R. A Genome Assembly of the Barley ‘Transformation Reference’ Cultivar Golden Promise. G3 Genes Genomes Genet. 2020, 10, 1823–1827. [Google Scholar] [CrossRef]
- Zhou, S.; Jiang, W.; Zhao, Y.; Zhou, D.X. Single-Cell Three-Dimensional Genome Structures of Rice Gametes and Unicellular Zygotes. Nat. Plants 2019, 5, 795–800. [Google Scholar] [CrossRef]
- Padmarasu, S.; Himmelbach, A.; Mascher, M.; Stein, N. In Situ Hi-C for Plants: An Improved Method to Detect Long-Range Chromatin Interactions. Methods Mol. Biol. 2019, 1933, 441–472. [Google Scholar] [CrossRef]
- Kadlof, M.; Rozycka, J.; Plewczynski, D. Spring Model—Chromatin Modeling Tool Based on OpenMM. Methods 2020, 181–182, 62–69. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the International AAAI Conference on Web and Social Media, San Jose, CA, USA, 17–20 May 2009; Volume 3, pp. 361–362. [Google Scholar] [CrossRef]
- Andrei Kashcha. Beautiful Graphs: Graph Data Structure in JavaScript. Available online: https://github.com/anvaka/ngraph.graph (accessed on 13 December 2020).
- Sepsi, A.; Fábián, A.; Jäger, K.; Heslop-Harrison, J.S.; Schwarzacher, T. Immunofish: Simultaneous Visualisation of Proteins and DNA Sequences Gives Insight into Meiotic Processes in Nuclei of Grasses. Front. Plant Sci. 2018, 9, 1193. [Google Scholar] [CrossRef]
- Hudakova, S.; Michalek, W.; Presting, G.G.; ten Hoopen, R.; dos Santos, K.; Jasencakova, Z.; Schubert, I. Sequence Organization of Barley Centromeres. Nucleic Acids Res. 2001, 29, 5029–5035. [Google Scholar] [CrossRef]
- Schwarzacher, T.; Heslop-Harrison, J.S. In Situ Hybridization to Plant Telomeres Using Synthetic Oligomers. Genome 1991, 34, 317–323. [Google Scholar] [CrossRef]
- Lenykó-Thegze, A.; Fábián, A.; Mihók, E.; Makai, D.; Cseh, A.; Sepsi, A. Pericentromeric Chromatin Reorganisation Follows the Initiation of Recombination and Coincides with Early Events of Synapsis in Cereals. Plant J. 2021, 107, 1585–1602. [Google Scholar] [CrossRef] [PubMed]
- Sepsi, A.; Higgins, J.D.; Heslop-Harrison, J.S.P.; Schwarzacher, T. CENH3 Morphogenesis Reveals Dynamic Centromere Associations during Synaptonemal Complex Formation and the Progression through Male Meiosis in Hexaploid Wheat. Plant J. 2017, 89, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Carstens, S.; Nilges, M.; Habeck, M. Bayesian Inference of Chromatin Structure Ensembles from Population-Averaged Contact Data. Proc. Natl. Acad. Sci. USA 2020, 117, 7824–7830. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Brown, W.M.; Klavans, R.; Boyack, K.W. OpenOrd: An Open-Source Toolbox for Large Graph Layout. Vis. Data Anal. 2011, 7868, 6. [Google Scholar] [CrossRef]
- Rabl, C. Über Zelltheilung; Morphologisches Jahrbuch: Leipzig, Germany, 1885; Volume 10. [Google Scholar]
- Anamthawat-Jonsson, K.; Heslop-Harrison, J.S. Centromeres, Telomeres and Chromatin in the Interphase Nucleus of Cereals. Caryologia 1990, 43, 205–213. [Google Scholar] [CrossRef]
- Galitsyna, A.A.; Gelfand, M.S. Single-Cell Hi-C Data Analysis: Safety in Numbers. Brief. Bioinform. 2021, 22, bbab316. [Google Scholar] [CrossRef]
- Vasquez, P.A.; Hult, C.; Adalsteinsson, D.; Lawrimore, J.; Forest, M.G.; Bloom, K. Entropy Gives Rise to Topologically Associating Domains. Nucleic Acids Res. 2016, 44, 5540–5549. [Google Scholar] [CrossRef]
- de Nooijer, S.; Wellink, J.; Mulder, B.; Bisseling, T. Non-Specific Interactions Are Sufficient to Explain the Position of Heterochromatic Chromocenters and Nucleoli in Interphase Nuclei. Nucleic Acids Res. 2009, 37, 3558–3568. [Google Scholar] [CrossRef]
- Robaszkiewicz, E.; Idziak-Helmcke, D.; Tkacz, M.A.; Chrominski, K.; Hasterok, R. The Arrangement of Brachypodium Distachyon Chromosomes in Interphase Nuclei. J. Exp. Bot. 2016, 67, 5571–5583. [Google Scholar] [CrossRef]
- Tiang, C.-L.; He, Y.; Pawlowski, W.P. Chromosome Organization and Dynamics during Interphase, Mitosis, and Meiosis in Plants. Plant Physiol. 2012, 158, 26–34. [Google Scholar] [CrossRef]
- Němečková, A.; Koláčková, V.; Vrána, J.; Doležel, J.; Hřibová, E. DNA Replication and Chromosome Positioning throughout the Interphase in Three-Dimensional Space of Plant Nuclei. J. Exp. Bot. 2020, 71, 6262–6272. [Google Scholar] [CrossRef] [PubMed]
- Arumuganathan, K.; Earle, E.D. Nuclear DNA Content of Some Important Plant Species. Plant Mol. Biol. Report. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Santos, A.P.; Shaw, P. Interphase Chromosomes and the Rabl Configuration: Does Genome Size Matter? J. Microsc. 2004, 214, 201–206. [Google Scholar] [CrossRef]
- Dong, F.; Jiang, J. Non-Rabl Patterns of Centromere and Telomere Distribution in the Interphase Nuclei of Plant Cells. Chromosome Res. 1998, 6, 551–558. [Google Scholar] [CrossRef]
- Fal, K.; Asnacios, A.; Chabouté, M.E.; Hamant, O. Nuclear Envelope: A New Frontier in Plant Mechanosensing? Biophys. Rev. 2017, 9, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Kirby, T.J.; Lammerding, J. Emerging Views of the Nucleus as a Cellular Mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef]
- Gaudet, P.; Dessimoz, C. Gene Ontology: Pitfalls, Biases, and Remedies. Methods Mol. Biol. 2017, 1446, 189–205. [Google Scholar] [CrossRef]
- Lajoie, B.R.; Dekker, J.; Kaplan, N. The Hitchhiker’s Guide to Hi-C Analysis: Practical Guidelines. Methods 2015, 72, 65–75. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makai, D.; Cseh, A.; Sepsi, A.; Makai, S. A Multigraph-Based Representation of Hi-C Data. Genes 2022, 13, 2189. https://doi.org/10.3390/genes13122189
Makai D, Cseh A, Sepsi A, Makai S. A Multigraph-Based Representation of Hi-C Data. Genes. 2022; 13(12):2189. https://doi.org/10.3390/genes13122189
Chicago/Turabian StyleMakai, Diána, András Cseh, Adél Sepsi, and Szabolcs Makai. 2022. "A Multigraph-Based Representation of Hi-C Data" Genes 13, no. 12: 2189. https://doi.org/10.3390/genes13122189
APA StyleMakai, D., Cseh, A., Sepsi, A., & Makai, S. (2022). A Multigraph-Based Representation of Hi-C Data. Genes, 13(12), 2189. https://doi.org/10.3390/genes13122189