
Enriched Pathways of Calcium Regulation, Cellular/Oxidative Stress, Inflammation, and Cell Proliferation Characterize Gluteal Muscle of Standardbred Horses between Episodes of Recurrent Exertional Rhabdomyolysis

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. RER-Susceptible Horses
2.2. Sample Collection
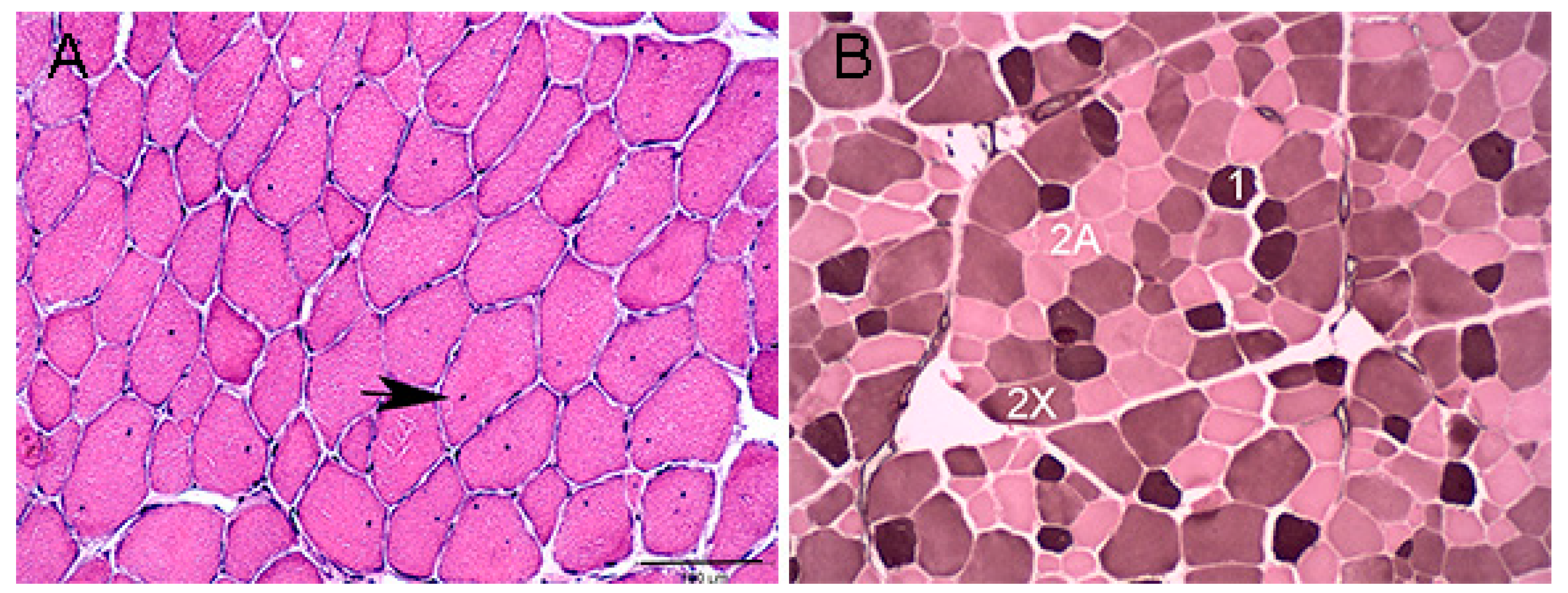
2.3. Muscle Histochemistry
2.4. Analyses of Signalment, Plasma CK, AST and Fiber Composition
2.5. Transcriptomics
2.5.1. RNA Extraction and Sequencing
2.5.2. Mapping and Assembling
2.5.3. RNA-Seq Count Normalization and Transformation
2.5.4. Differential Gene Expression Analysis
2.6. Proteomics
2.6.1. Sample Preparation and LC/MS/MS
2.6.2. Quantitative Data Analysis
2.7. Transcriptome and Proteome Co-Inertia Analysis
2.8. Enrichment and Pathway Analysis
2.9. Muscle Biochemistry
2.9.1. Reactive Oxygen Species (ROS)
2.9.2. Coenzyme Q10
2.9.3. Glutathione
2.9.4. Statistical Analyses of ROS, Glutathione and Coenzyme Q10
3. Results
3.1. Horses and Muscle Histochemistry
3.2. Transcriptomics
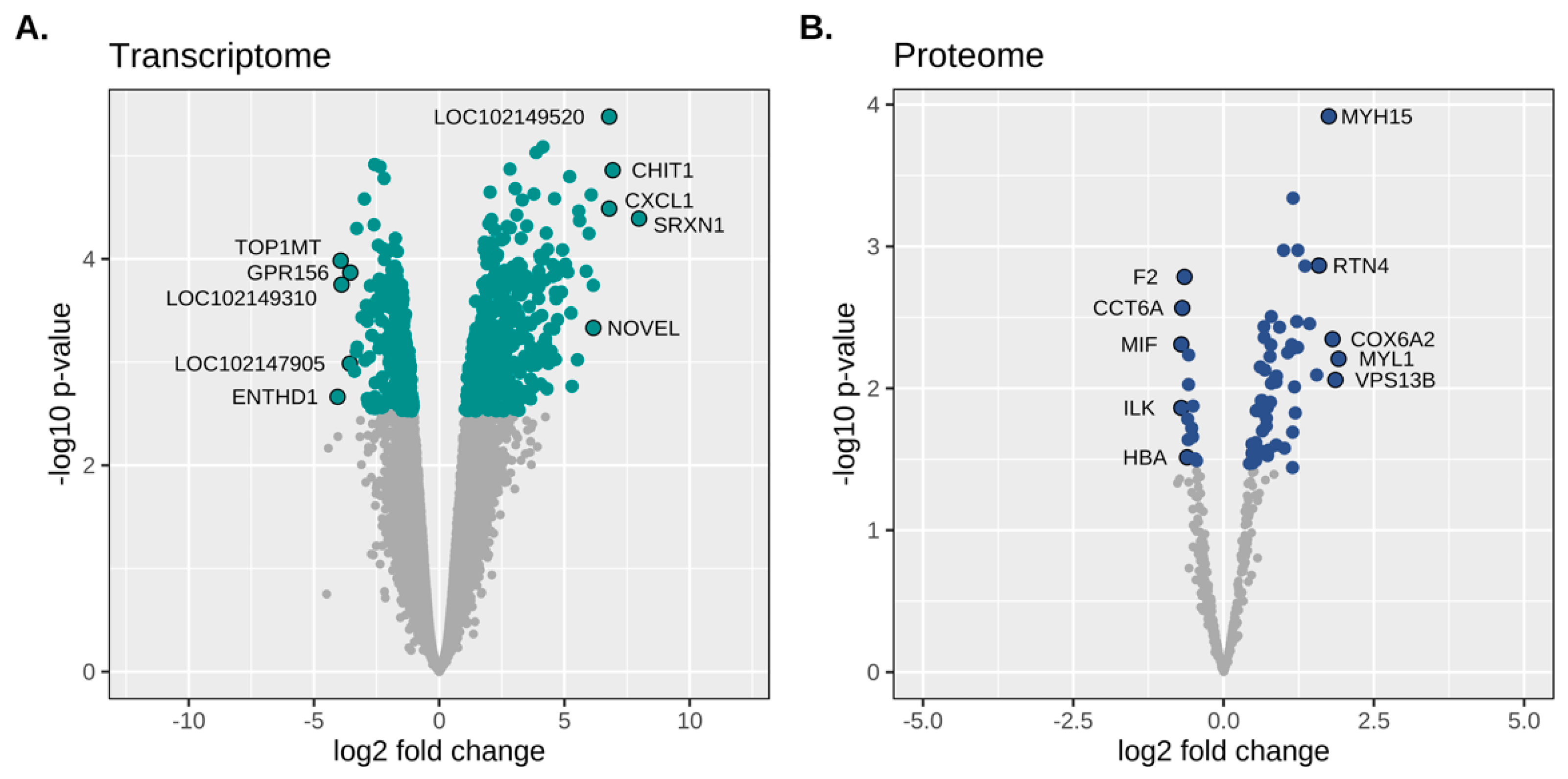
3.2.1. Expressed Gene Transcripts
3.2.2. Gene Ontology Pathway for RER Transcriptomic Analysis
3.3. Proteomics
3.3.1. Confident Protein Classification, Differential Expression and GO Analysis
3.3.2. Gene Ontology (GO) Pathway for RER Proteomic Analysis
3.3.3. Differentially Expressed Genes with Differentially Expressed Proteins
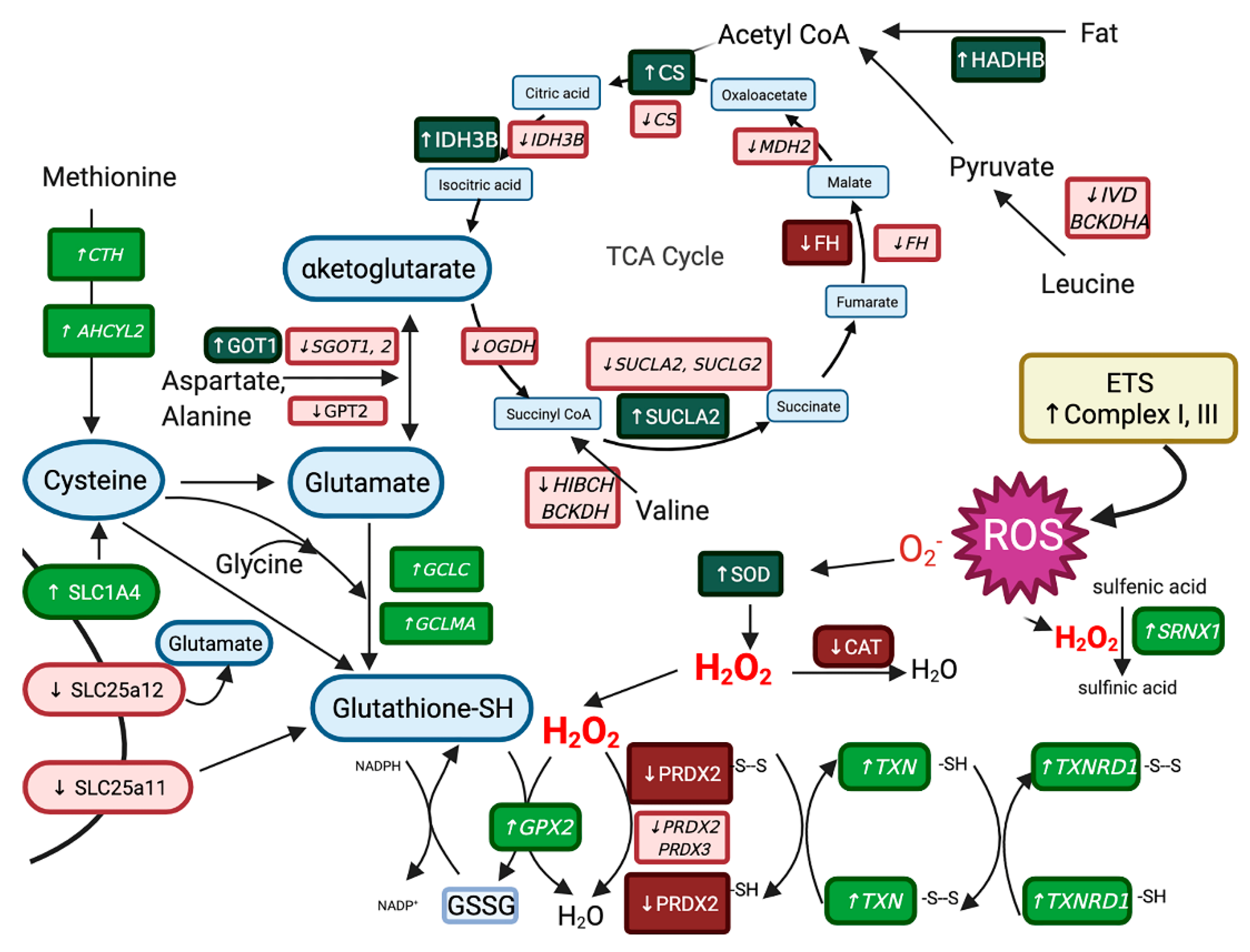
3.4. Reactome for Merged Differentially Expressed Genes and Proteins
3.5. Co-Inertia Analysis
3.6. Muscle Biochemistry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Isgren, C.M.; Upjohn, M.M.; Fernandez-Fuente, M.; Massey, C.; Pollott, G.; Verheyen, K.L.; Piercy, R.J. Epidemiology of exertional rhabdomyolysis susceptibility in standardbred horses reveals associated risk factors and underlying enhanced performance. PLoS ONE 2010, 5, e11594. [Google Scholar] [CrossRef] [PubMed]
- MacLeay, J.M.; Sorum, S.A.; Valberg, S.J.; Marsh, W.E.; Sorum, M.D. Epidemiologic analysis of factors influencing exertional rhabdomyolysis in Thoroughbreds. Am. J. Vet. Res. 1999, 60, 1562–1566. [Google Scholar] [PubMed]
- McGowan, C.M.; Fordham, T.; Christley, R.M. Incidence and risk factors for exertional rhabdomyolysis in thoroughbred racehorses in the United Kingdom. Vet. Rec. 2002, 151, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Norton, E.M.; Mickelson, J.R.; Binns, M.M.; Blott, S.C.; Caputo, P.; Isgren, C.M.; McCoy, A.M.; Moore, A.; Piercy, R.J.; Swinburne, J.E.; et al. Heritability of Recurrent Exertional Rhabdomyolysis in Standardbred and Thoroughbred Racehorses Derived From SNP Genotyping Data. J. Hered. 2016, 107, 537–543. [Google Scholar] [CrossRef]
- Fritz, K.L.; McCue, M.E.; Valberg, S.J.; Rendahl, A.K.; Mickelson, J.R. Genetic mapping of recurrent exertional rhabdomyolysis in a population of North American Thoroughbreds. Anim. Genet. 2012, 43, 730–738. [Google Scholar] [CrossRef]
- Dranchak, P.K.; Valberg, S.J.; Onan, G.W.; Gallant, E.M.; MacLeay, J.M.; McKenzie, E.C.; De La Corte, F.D.; Ekenstedt, K.; Mickelson, J.R. Inheritance of recurrent exertional rhabdomyolysis in thoroughbreds. J. Am. Vet. Med. Assoc. 2005, 227, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, K.; Velez-Irizarry, D.; Fenger, C.; Schott, M.; Valberg, S.J. Pathways of calcium regulation, electron transport, and mitochondrial protein translation are molecular signatures of susceptibility to recurrent exertional rhabdomyolysis in Thoroughbred racehorses. PLoS ONE 2021, 16, e0244556. [Google Scholar] [CrossRef] [PubMed]
- Barrey, E.; Bonnamy, B.; Barrey, E.J.; Mata, X.; Chaffaux, S.; Guerin, G. Muscular microRNA expressions in healthy and myopathic horses suffering from polysaccharide storage myopathy or recurrent exertional rhabdomyolysis. Equine Vet. J. 2010, 42, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Barrey, E.; Jayr, L.; Mucher, E.; Gospodnetic, S.; Joly, F.; Benech, P.; Alibert, O.; Gidrol, X.; Mata, X.; Vaiman, A.; et al. Transcriptome analysis of muscle in horses suffering from recurrent exertional rhabdomyolysis revealed energetic pathway alterations and disruption in the cytosolic calcium regulation. Anim. Genet. 2012, 43, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Scholte, H.R.; Verduin, M.H.; Ross, J.D.; Van den Hoven, R.; Wensing, T.; Breuking, H.J.; Meijer, A.E. Equine exertional rhabdomyolysis: Activity of the mitochondrial respiratory chain and the carnitine system in skeletal muscle. Equine Vet. J. 1991, 23, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Valberg, S.; Haggendal, J.; Lindholm, A. Blood chemistry and skeletal muscle metabolic responses to exercise in horses with recurrent exertional rhabdomyolysis. Equine Vet. J. 1993, 25, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Valberg, S.; Jonsson, L.; Lindholm, A.; Holmgren, N. Muscle histopathology and plasma aspartate aminotransferase, creatine kinase and myoglobin changes with exercise in horses with recurrent exertional rhabdomyolysis. Equine Vet. J. 1993, 25, 11–16. [Google Scholar] [CrossRef]
- Van den Hoven, R.; Breukink, H.J.; Wensing, T.; Meijer, A.E.; Tigges, A.J. Loosely coupled skeletal muscle mitochondria in exertional rhabdomyolysis. Equine Vet. J. 1986, 18, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Beech, J.; Lindborg, S.; Fletcher, J.E.; Lizzo, F.; Tripolitis, L.; Braund, K. Caffeine contractures, twitch characteristics and the threshold for Ca2+-induced Ca2+ release in skeletal muscle from horses with chronic intermittent rhabdomyolysis. Res. Vet. Sci. 1993, 54, 110–117. [Google Scholar] [CrossRef]
- Houben, R.; Leleu, C.; Fraipont, A.; Serteyn, D.; Votion, D.M. Determination of muscle mitochondrial respiratory capacity in Standardbred racehorses as an aid to predicting exertional rhabdomyolysis. Mitochondrion 2015, 24, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; De Palma, S.; Vigano, A.; Lochmuller, H.; Muntoni, F.; Ferlini, A.; Mora, M.; et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: Changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexia Sarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, A.; Piehl, K. Fibre composition, enzyme activity and concentrations of metabolites and electrolytes in muscles of standardbred horses. Acta Vet. Scand. 1974, 15, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Cumming, J.K.F.; Mahon, M. Color Atlas of Muscle Pathology; Mosby-Wolfe: London, UK, 1994. [Google Scholar]
- Valberg, S.J.; Nicholson, A.M.; Lewis, S.S.; Reardon, R.A.; Finno, C.J. Clinical and histopathological features of myofibrillar myopathy in Warmblood horses. Equine Vet. J. 2017, 49, 739–745. [Google Scholar] [CrossRef]
- Blomstrand, E.; Ekblom, B. The needle biopsy technique for fibre type determination in human skeletal muscle—A methodological study. Acta Physiol. Scand. 1982, 116, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Williams, Z.J.; Velez-Irizarry, D.; Petersen, J.L.; Ochala, J.; Finno, C.J.; Valberg, S.J. Candidate gene expression and coding sequence variants in Warmblood horses with myofibrillar myopathy. Equine Vet. J. 2021, 53, 306–315. [Google Scholar] [CrossRef]
- Smeds, L.; Künstner, A. ConDeTri-a content dependent read trimmer for Illumina data. PLoS ONE 2011, 6, e26314. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Liu, R.; Holik, A.Z.; Su, S.; Jansz, N.; Chen, K.; Leong, H.S.; Blewitt, M.E.; Asselin-Labat, M.L.; Smyth, G.K.; Ritchie, M.E. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 2015, 43, e97. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Smyth, G.K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2009, 25, 765–771. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Branson, O.E.; Freitas, M.A. A multi-model statistical approach for proteomic spectral count quantitation. J. Proteom. 2016, 144, 23–32. [Google Scholar] [CrossRef]
- Min, E.J.; Safo, S.E.; Long, Q. Penalized co-inertia analysis with applications to—Omics data. Bioinformatics 2019, 35, 1018–1025. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef]
- Carlson, M. Genome Wide Annotation for Human, R package version 3.8.2; Bioconductor, 2019. [CrossRef]
- Farries, G.; Bryan, K.; McGivney, C.L.; McGettigan, P.A.; Gough, K.F.; Browne, J.A.; MacHugh, D.E.; Katz, L.M.; Hill, E.W. Expression Quantitative Trait Loci in Equine Skeletal Muscle Reveals Heritable Variation in Metabolism and the Training Responsive Transcriptome. Front. Genet. 2019, 10, 1215. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Pandey, R.; Riley, C.L.; Mills, E.M.; Tiziani, S. Highly sensitive and selective determination of redox states of coenzymes Q9 and Q10 in mice tissues: Application of orbitrap mass spectrometry. Anal Chim Acta 2018, 1011, 68–76. [Google Scholar] [CrossRef]
- Chen, Z.; Putt, D.A.; Lash, L.H. Enrichment and functional reconstitution of glutathione transport activity from rabbit kidney mitochondria: Further evidence for the role of the dicarboxylate and 2-oxoglutarate carriers in mitochondrial glutathione transport. Arch Biochem. Biophys. 2000, 373, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Witherspoon, J.W.; Meilleur, K.G. Review of RyR1 pathway and associated pathomechanisms. Acta Neuropathol. Commun. 2016, 4, 121. [Google Scholar] [CrossRef]
- Munro, M.L.; Jayasinghe, I.D.; Wang, Q.; Quick, A.; Wang, W.; Baddeley, D.; Wehrens, X.H.; Soeller, C. Junctophilin-2 in the nanoscale organisation and functional signalling of ryanodine receptor clusters in cardiomyocytes. J. Cell Sci. 2016, 129, 4388–4398. [Google Scholar] [CrossRef]
- Arcuri, C.; Giambanco, I.; Bianchi, R.; Donato, R. Subcellular localization of S100A11 (S100C, calgizzarin) in developing and adult avian skeletal muscles. Biochim. Biophys. Acta 2002, 1600, 84–94. [Google Scholar] [CrossRef]
- Lee, K.J.; Hyun, C.; Woo, J.S.; Park, C.S.; Kim, D.H.; Lee, E.H. Stromal interaction molecule 1 (STIM1) regulates sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 1a (SERCA1a) in skeletal muscle. Pflug. Arch. 2014, 466, 987–1001. [Google Scholar] [CrossRef]
- Valberg, S.J.; Soave, K.; Williams, Z.J.; Perumbakkam, S.; Schott, M.; Finno, C.J.; Petersen, J.L.; Fenger, C.; Autry, J.M.; Thomas, D.D. Coding sequences of sarcoplasmic reticulum calcium ATPase regulatory peptides and expression of calcium regulatory genes in recurrent exertional rhabdomyolysis. J. Vet. Intern. Med. 2019, 33, 933–941. [Google Scholar] [CrossRef]
- Cheng, A.J.; Andersson, D.C.; Lanner, J.T. Can’t live with or without it: Calcium and its role in Duchenne muscular dystrophy-induced muscle weakness. Focus on “SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models”. Am. J. Physiol. Cell Physiol. 2015, 308, C697–C698. [Google Scholar] [CrossRef]
- Valberg, S.; Essen Gustavsson, B.; Skoglund Wallberg, H. Oxidative capacity of skeletal muscle fibres in racehorses: Histochemical versus biochemical analysis. Equine Vet. J. 1988, 20, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Roneus, M.; Essen-Gustavsson, B.; Lindholm, A.; Persson, S.G. Skeletal muscle characteristics in young trained and untrained standardbred trotters. Equine Vet. J. 1992, 24, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Roneus, N.; Essen-Gustavsson, B.; Lindholm, A.; Eriksson, Y. Plasma lactate response to submaximal and maximal exercise tests with training, and its relationship to performance and muscle characteristics in standardbred trotters. Equine Vet. J. 1994, 26, 117–121. [Google Scholar] [CrossRef]
- Valberg, S. Muscling in on the cause of tying up. In Proceedings of the 58th Annual Convention of the American Association of Equine Practitioners, Anaheim, CA, USA, 1–5 December 2012; pp. 85–123. [Google Scholar]
- Weins, A.; Schwarz, K.; Faul, C.; Barisoni, L.; Linke, W.A.; Mundel, P. Differentiation- and stress-dependent nuclear cytoplasmic redistribution of myopodin, a novel actin-bundling protein. J. Cell Biol. 2001, 155, 393–404. [Google Scholar] [CrossRef]
- Frank, D.; Frey, N. Cardiac Z-disc signaling network. J. Biol. Chem. 2011, 286, 9897–9904. [Google Scholar] [CrossRef]
- Kostek, M.C.; Chen, Y.W.; Cuthbertson, D.J.; Shi, R.; Fedele, M.J.; Esser, K.A.; Rennie, M.J. Gene expression responses over 24 h to lengthening and shortening contractions in human muscle: Major changes in CSRP3, MUSTN1, SIX1, and FBXO32. Physiol. Genom. 2007, 31, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.; Gallego-Villar, L.; Rivera-Barahona, A.; Oyarzabal, A.; Perez, B.; Rodriguez-Pombo, P.; Desviat, L.R. Altered Redox Homeostasis in Branched-Chain Amino Acid Disorders, Organic Acidurias, and Homocystinuria. Oxid. Med. Cell. Longev. 2018, 2018, 1246069. [Google Scholar] [CrossRef]
- Kinnunen, S.; Hyyppa, S.; Lappalainen, J.; Oksala, N.; Venojarvi, M.; Nakao, C.; Hanninen, O.; Sen, C.K.; Atalay, M. Exercise-induced oxidative stress and muscle stress protein responses in trotters. Eur. J. Appl. Physiol. 2005, 93, 496–501. [Google Scholar] [CrossRef]
- Bruce, M.; Constantin-Teodosiu, D.; Greenhaff, P.L.; Boobis, L.H.; Williams, C.; Bowtell, J.L. Glutamine supplementation promotes anaplerosis but not oxidative energy delivery in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E669–E675. [Google Scholar] [CrossRef]
- Peterson, J.M.; Bakkar, N.; Guttridge, D.C. NF-kappaB signaling in skeletal muscle health and disease. Curr. Top. Dev. Biol. 2011, 96, 85–119. [Google Scholar] [CrossRef]
- Di Rosa, M.; Malaguarnera, G.; De Gregorio, C.; Drago, F.; Malaguarnera, L. Evaluation of CHI3L-1 and CHIT-1 expression in differentiated and polarized macrophages. Inflammation 2013, 36, 482–492. [Google Scholar] [CrossRef]
- Lombardo, S.D.; Mazzon, E.; Mangano, K.; Basile, M.S.; Cavalli, E.; Mammana, S.; Fagone, P.; Nicoletti, F.; Petralia, M.C. Transcriptomic Analysis Reveals Involvement of the Macrophage Migration Inhibitory Factor Gene Network in Duchenne Muscular Dystrophy. Genes 2019, 10, 939. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.M.A.; Elfadl, A.K.; Park, S.; Kim, Y.D.; Chung, M.J.; Son, J.Y.; Yun, H.H.; Park, J.M.; Yim, J.H.; Jung, S.J.; et al. Nogo-A Is Critical for Pro-Inflammatory Gene Regulation in Myocytes and Macrophages. Cells 2021, 10, 282. [Google Scholar] [CrossRef]
- Ciciliot, S.; Schiaffino, S. Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des. 2010, 16, 906–914. [Google Scholar] [CrossRef]
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [PubMed]
- Folker, E.S.; Baylies, M.K. Nuclear positioning in muscle development and disease. Front. Physiol. 2013, 4, 363. [Google Scholar] [CrossRef] [PubMed]
- Roman, W.; Gomes, E.R. Nuclear positioning in skeletal muscle. Semin. Cell Dev. Biol. 2018, 82, 51–56. [Google Scholar] [CrossRef]
- Valberg, S.J.; Mickelson, J.R.; Gallant, E.M.; MacLeay, J.M.; Lentz, L.; de la Corte, F. Exertional rhabdomyolysis in quarter horses and thoroughbreds: One syndrome, multiple aetiologies. Equine Vet. J. 1999, 31, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, D.; Khalique, U.; Belanto, J.J.; Kenea, A.; Talsness, D.M.; Olthoff, J.T.; Tran, M.D.; Zaal, K.J.; Pak, K.; Pinal-Fernandez, I.; et al. Persistent upregulation of the beta-tubulin tubb6, linked to muscle regeneration, is a source of microtubule disorganization in dystrophic muscle. Hum. Mol. Genet. 2019, 28, 1117–1135. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, K.; Qian, P.Y. Proteomics: Challenges, techniques and possibilities to overcome biological sample complexity. Hum. Genom. Proteom. 2009, 2009, 239204. [Google Scholar] [CrossRef] [PubMed]
- McGivney, B.A.; Eivers, S.S.; MacHugh, D.E.; MacLeod, J.N.; O’Gorman, G.M.; Park, S.D.; Katz, L.M.; Hill, E.W. Transcriptional adaptations following exercise in thoroughbred horse skeletal muscle highlights molecular mechanisms that lead to muscle hypertrophy. BMC Genom. 2009, 10, 638. [Google Scholar] [CrossRef]
- Van Pelt, D.W.; Kharaz, Y.A.; Sarver, D.C.; Eckhardt, L.R.; Dzierzawski, J.T.; Disser, N.P.; Piacentini, A.N.; Comerford, E.; McDonagh, B.; Mendias, C.L. Multiomics analysis of the mdx/mTR mouse model of Duchenne muscular dystrophy. Connect. Tissue Res. 2021, 62, 24–39. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Age | Time | CK | AST | Type 1 | Type 2A | Type 2X | |
|---|---|---|---|---|---|---|---|---|
| Yrs | Hours | U/L 1 | U/L 2 | % | % | % | ||
| Control | 7 | 3.9 ± 1.3 | 1.8 ± 0.6 | 257 ± 95 | 371 ± 96 | 10.8 ± 1.7 | 39.3 ± 9.4 | 49.8 ± 10.0 |
| RER-susceptible | 9 | 4.8 ± 1.6 | 1.7 ± 0.7 | 433 ± 275 | 644 ± 564 | 16.9 ± 4.4 | 45.3 ± 13.9 | 37.4 ± 13.9 |
| p value | 0.22 | 0.74 | 0.14 | 0.17 | 0.007 | 0.37 | 0.08 |
| Category for GO Term | Total Number Genes | Number of GO Terms | Genes/GO Grouped Category |
| Upregulated DEG | 385 | ||
| Inflammation/Immune response | 62 | 5–49 | |
| Cell proliferation | 31 | 5–22 | |
| Cell/oxidative stress | 31 | 7–30 | |
| Protein modification | 7 | 13–28 | |
| Extracellular matrix | 7 | 6–26 | |
| Regulatory processes | 6 | 4–23 | |
| Amino acid/amine metabolism | 5 | 13–20 | |
| Mitochondrial membranes | 3 | 5–10 | |
| Ketone metabolism | 2 | 14–16 | |
| Response to stimulus | 2 | 19–28 | |
| Downregulated DEG | 305 | ||
| Calcium ion regulation (CACNA2D1/CASQ) | 39 | 4–13 | |
| Purine nucleotide metabolism | 32 | 5–42 | |
| Electron transport | 29 | 4–54 | |
| Other metabolism | 23 | 3–75 | |
| Amino acid metabolism | 15 | 3–20 | |
| Fat metabolism | 10 | 4–20 | |
| Carbohydrate metabolism | 10 | 5–28 | |
| Tricarboxylic acid cycle | 9 | 4–19 | |
| Polysaccharide metabolism | 9 | 5–28 | |
| Thermogenesis | 5 | 8–13 | |
| Hormone secretion | 5 | 9–16 | |
| Other Ion transport | 4 | 4–28 | |
| Thioesters | 3 | 9–20 | |
| Signaling | 3 | 3–15 | |
| Actin | 1 | 5 | |
| Reactome All DEG and DEP | Total Number Genes | Number of RH Terms | Genes/RH Grouped Category |
| 498 | |||
| Cell proliferation | 45 | 11–18 | |
| Immune response | 11 | 15–40 | |
| NFkB Immune response | 7 | 14–17 | |
| Cell/oxidative stress | 8 | 10–43 | |
| Protein processing | 8 | 7–20 | |
| Transcription | 7 | 12–19 | |
| Metabolism | 9 | 5–47 | |
| Electron transport | 7 | 11–48 | |
| TCA cycle | 5 | 5–69 | |
| Extracellular matrix | 5 | 5–13 | |
| Signaling | 4 | 4–14 | |
| Transporters | 3 | 12–14 | |
| Muscle contraction | 2 | 9–19 |
| Gene ID | Protein Name | Log2FC | Adj. P |
|---|---|---|---|
| Mitochondria | |||
| Complex I | |||
| NDUFB7 | Cluster of NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 7 | 1.23 | 4.75 × 10−2 |
| NDUFA2 | Cluster of NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 2 | 0.71 | 7.20 × 10−2 |
| NDUFS2 * | NADH dehydrogenase [ubiquinone] iron-sulfur protein 2 | 0.47 | 8.84 × 10−2 |
| Complex III | |||
| UQCRB | cytochrome b-c1 complex subunit 7 | 1.55 | 5.20 × 10−2 |
| LOC111767815 | cytochrome b-c1 complex subunit 8 | 1.35 | 4.56 × 10−2 |
| LOC100053634 | cytochrome b-c1 complex subunit Rieske | 0.54 | 6.19 × 10−2 |
| UQCRC1 | Cluster of cytochrome b-c1 complex subunit 1 | 0.64 | 6.19 × 10−2 |
| LOC100051878 | Cluster of cytochrome b-c1 complex subunit 6 | −0.45 | 9.21 × 10−2 |
| Complex IV | |||
| LOC100055813 | cytochrome c oxidase subunit 6C | 1.86 | 5.20 × 10−2 |
| COX6A2 | cytochrome c oxidase subunit 6A2 | 1.81 | 4.75 × 10−2 |
| CYC1 * | cytochrome c1 heme protein | 0.67 | 4.75 × 10−2 |
| Complex V | |||
| ATP5MG | ATP synthase subunit g | 1.24 | 4.56 × 10−2 |
| ATP5PF * | ATP synthase-coupling factor 6 | 1.07 | 4.75 × 10−2 |
| ATP5F1D | ATP synthase subunit delta | 0.73 | 9.00 × 10−2 |
| ATP5F1B | ATP synthase subunit beta | 0.43 | 9.39 × 10−2 |
| SLC25A4 * | ADP/ATP translocase 1 | 0.52 | 8.84 × 10−2 |
| Fat metabolism | |||
| HADHB | trifunctional enzyme subunit beta | 1.22 | 4.75 × 10−2 |
| TCA cycle | |||
| CS * | citrate synthase | 0.61 | 5.20 × 10−2 |
| SUCLA2 * | Succinate—CoA ligase subunit beta | 0.88 | 5.20 × 10−2 |
| IDH3B * | isocitrate dehydrogenase [NAD] subunit beta isoform X2 | 0.77 | 4.75 × 10−2 |
| FH * | fumarate hydratase | −0.51 | 6.19 × 10−2 |
| Other mitochondrial | |||
| SOD2 | superoxide dismutase [Mn] mitochondrial precursor | 1.15 | 4.75 × 10−2 |
| VDAC2 | voltage-dependent anion-selective channel protein 2 | 0.85 | 5.20 × 10−2 |
| PPA2 * | Cluster of inorganic pyrophosphatase 2 | 0.53 | 9.21 × 10−2 |
| MRPS36 * | 28S ribosomal protein S36 | 0.54 | 8.43 × 10−2 |
| Protein Name | Log2FC | Adj. P | |
|---|---|---|---|
| Sarcomere | |||
| Thick and thin filaments | |||
| MYL1 | myosin light chain 1/3 skeletal muscle isoform isoform X1-fast-twitch | 1.91 | 4.75 × 10−2 |
| TNNI1 | troponin I slow-twitch | 1.43 | 4.75 × 10−2 |
| TPM3 | tropomyosin alpha-3 chain isoform X4 slow-twitch | 0.93 | 4.75 × 10−2 |
| LOC100062893 | myosin 8 | 0.79 | 5.20 × 10−2 |
| TPM1 | tropomyosin alpha-1 chain isoform X5 | 0.71 | 6.55 × 10−2 |
| MYOM1 | Cluster of myomesin-1 | 0.69 | 5.20 × 10−2 |
| MYL3 | myosin light chain 3-slow twitch | 0.67 | 6.19 × 10−2 |
| TNNI2 | troponin I fast twitch | 0.62 | 8.77 × 10−2 |
| MYBPH | myosin-binding protein H | −0.53 | 7.31 × 10−2 |
| Z disc | |||
| CSRP3 | cysteine and glycine-rich protein 3 | 1.16 | 4.55 × 10−2 |
| FHL1 | four and a half LIM domains protein 1 isoform X3 | 1.13 | 4.75 × 10−2 |
| MYOT | myotilin isoform X1 | 1.01 | 8.74 × 10−2 |
| PDLIM3 | Cluster of PDZ and LIM domain protein 3 | 1.00 | 4.56 × 10−2 |
| PDLIM5 | PDZ and LIM domain protein 5 isoform X9 | 0.87 | 5.20 × 10−2 |
| DES | desmin | 0.79 | 4.75 × 10−2 |
| MYOZ1 | myozenin-1 fast-twitch | 0.68 | 4.75 × 10−2 |
| SYNPO2 | synaptopodin-2 | 0.63 | 6.19 × 10−2 |
| Metabolism | |||
| Polysaccharides | |||
| AKR1B1 | aldose reductase | 1.19 | 6.19 × 10−2 |
| ENO2 | gamma-enolase | 1.18 | 5.26 × 10−2 |
| PGAM2 | phosphoglycerate mutase 2 | 0.75 | 8.74 × 10−2 |
| ALDOC | fructose-bisphosphate aldolase C | 0.63 | 6.19 × 10−2 |
| PYGM | myophosphorylase | 0.47 | 8.47 × 10−2 |
| GYG | glycogenin 1 | 0.55 | 8.74 × 10−2 |
| Protein | |||
| GOT1 * | aspartate aminotransferase cytoplasmic | 0.78 | 6.19 × 10−2 |
| PADI2 | protein-arginine deiminase type-2 | −0.60 | 6.55 × 10−2 |
| Lipid | |||
| PGP | glycerol-3-phosphate phosphatase | −0.48 | 9.18 × 10−2 |
| Antioxidant | |||
| PRDX2 * | Cluster of peroxiredoxin-2 | −0.58 | 5.20 × 10−2 |
| CAT | Cluster of catalase | −0.59 | 8.19 × 10−2 |
| Heat Shock factors | |||
| HSPA8 | heat shock cognate 71 kDa protein | 0.79 | 4.75 × 10−2 |
| HSPA1A | heat shock 70kDa protein 1A | 0.48 | 9.39 × 10−2 |
| Inflammation | |||
| RTN4 | reticulon-4 isoform X4 | 1.59 | 4.56 × 10−2 |
| MIF | macrophage migration inhibitory factor | −0.70 | 4.75 × 10−2 |
| Other | |||
| MYH15 | myosin-15 | 1.75 | 2.41 × 10−2 |
| AHSG | alpha-2-HS-glycoprotein | 1.15 | 9.88 × 10−2 |
| SPI2 | alpha-1-antiproteinase 2 isoform X1 | 1.15 | 7.51 × 10−2 |
| LOC100065068 | alpha-1-antiproteinase 2-like precursor | 0.88 | 8.47 × 10−2 |
| EEF1A2 | elongation factor 1-alpha 2 | 0.73 | 6.19 × 10−2 |
| PEBP1 | phosphatidylethanolamine-binding protein 1 | 0.65 | 7.50 × 10−2 |
| YWHAE | Cluster of 14-3-3 protein epsilon | 0.58 | 6.19 × 10−2 |
| EHD2 | EH domain-containing protein 2 | −0.52 | 7.97 × 10−2 |
| APOA1 | apolipoprotein A-I | −0.58 | 4.75 × 10−2 |
| HBA | hemoglobin subunit alpha | −0.61 | 9.11 × 10−2 |
| F2 | prothrombin | −0.65 | 4.66 × 10−2 |
| CCT6A | T-complex protein 1 subunit zeta | −0.69 | 4.75 × 10−2 |
| ILK | integrin-linked protein kinase isoform X2 | −0.70 | 6.19 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valberg, S.J.; Velez-Irizarry, D.; Williams, Z.J.; Henry, M.L.; Iglewski, H.; Herrick, K.; Fenger, C. Enriched Pathways of Calcium Regulation, Cellular/Oxidative Stress, Inflammation, and Cell Proliferation Characterize Gluteal Muscle of Standardbred Horses between Episodes of Recurrent Exertional Rhabdomyolysis. Genes 2022, 13, 1853. https://doi.org/10.3390/genes13101853
Valberg SJ, Velez-Irizarry D, Williams ZJ, Henry ML, Iglewski H, Herrick K, Fenger C. Enriched Pathways of Calcium Regulation, Cellular/Oxidative Stress, Inflammation, and Cell Proliferation Characterize Gluteal Muscle of Standardbred Horses between Episodes of Recurrent Exertional Rhabdomyolysis. Genes. 2022; 13(10):1853. https://doi.org/10.3390/genes13101853
Chicago/Turabian StyleValberg, Stephanie J., Deborah Velez-Irizarry, Zoë J. Williams, Marisa L. Henry, Hailey Iglewski, Keely Herrick, and Clara Fenger. 2022. "Enriched Pathways of Calcium Regulation, Cellular/Oxidative Stress, Inflammation, and Cell Proliferation Characterize Gluteal Muscle of Standardbred Horses between Episodes of Recurrent Exertional Rhabdomyolysis" Genes 13, no. 10: 1853. https://doi.org/10.3390/genes13101853
APA StyleValberg, S. J., Velez-Irizarry, D., Williams, Z. J., Henry, M. L., Iglewski, H., Herrick, K., & Fenger, C. (2022). Enriched Pathways of Calcium Regulation, Cellular/Oxidative Stress, Inflammation, and Cell Proliferation Characterize Gluteal Muscle of Standardbred Horses between Episodes of Recurrent Exertional Rhabdomyolysis. Genes, 13(10), 1853. https://doi.org/10.3390/genes13101853