Single Cell RNA-Seq Identifies Immune-Related Prognostic Model and Key Signature-SPP1 in Pancreatic Ductal Adenocarcinoma

 ,
,
Abstract
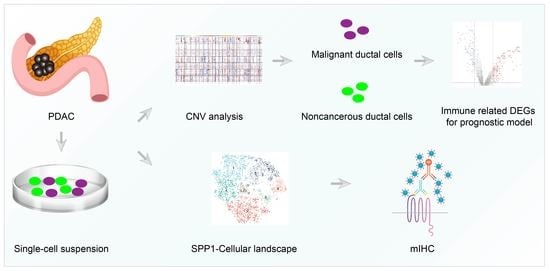
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patient Cohorts and Study Design
2.2. Single Cell RNA-Seq Analysis
2.3. Cell Culture
2.4. Development of Prognostic Model
2.5. Tumor Immune Infiltration Analysis
2.6. Multiple Color IHC
2.7. Statistical Analysis
3. Results
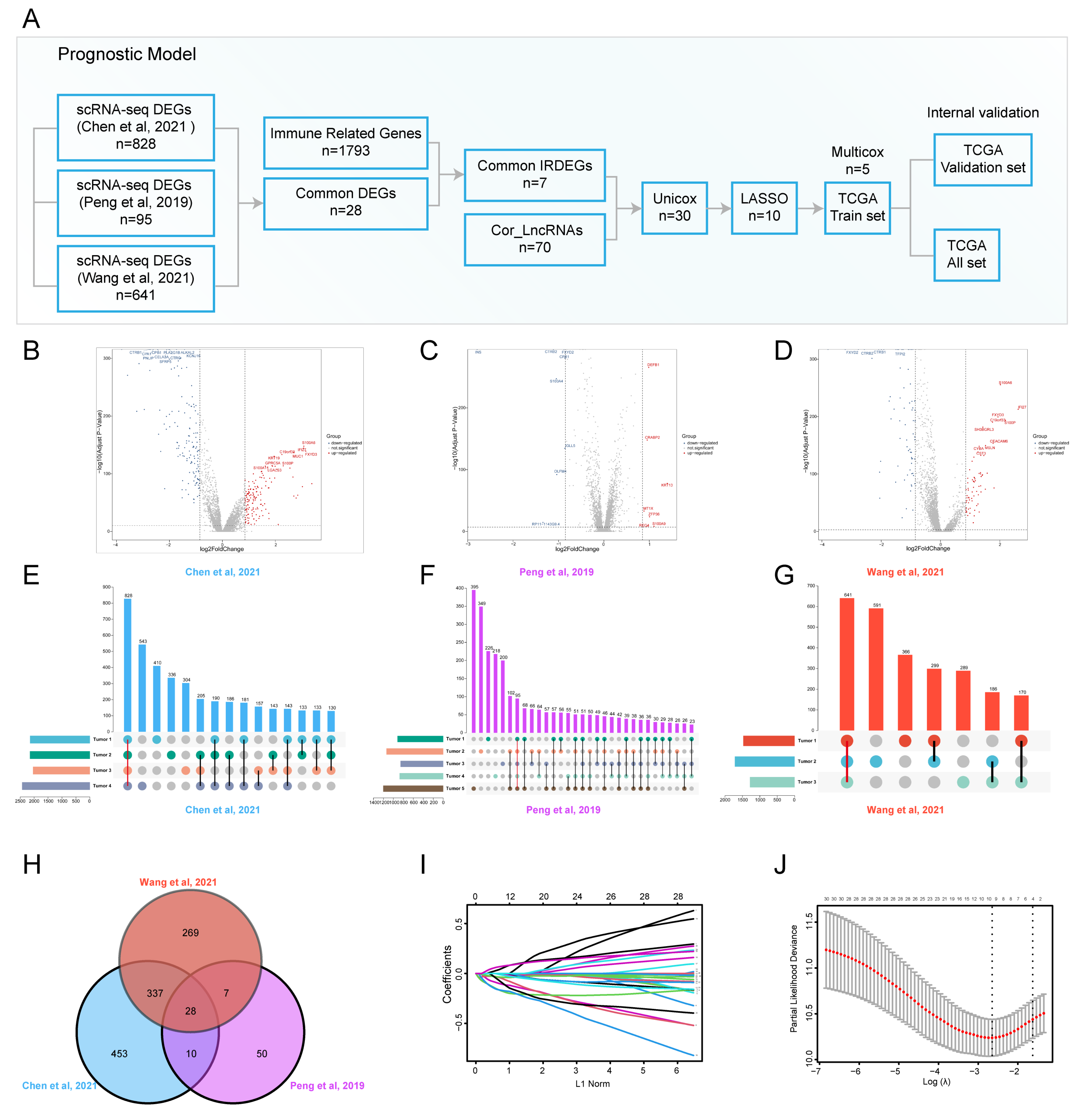
3.1. ScRNA-Seq Identified Immune Related DEGs for PDAC
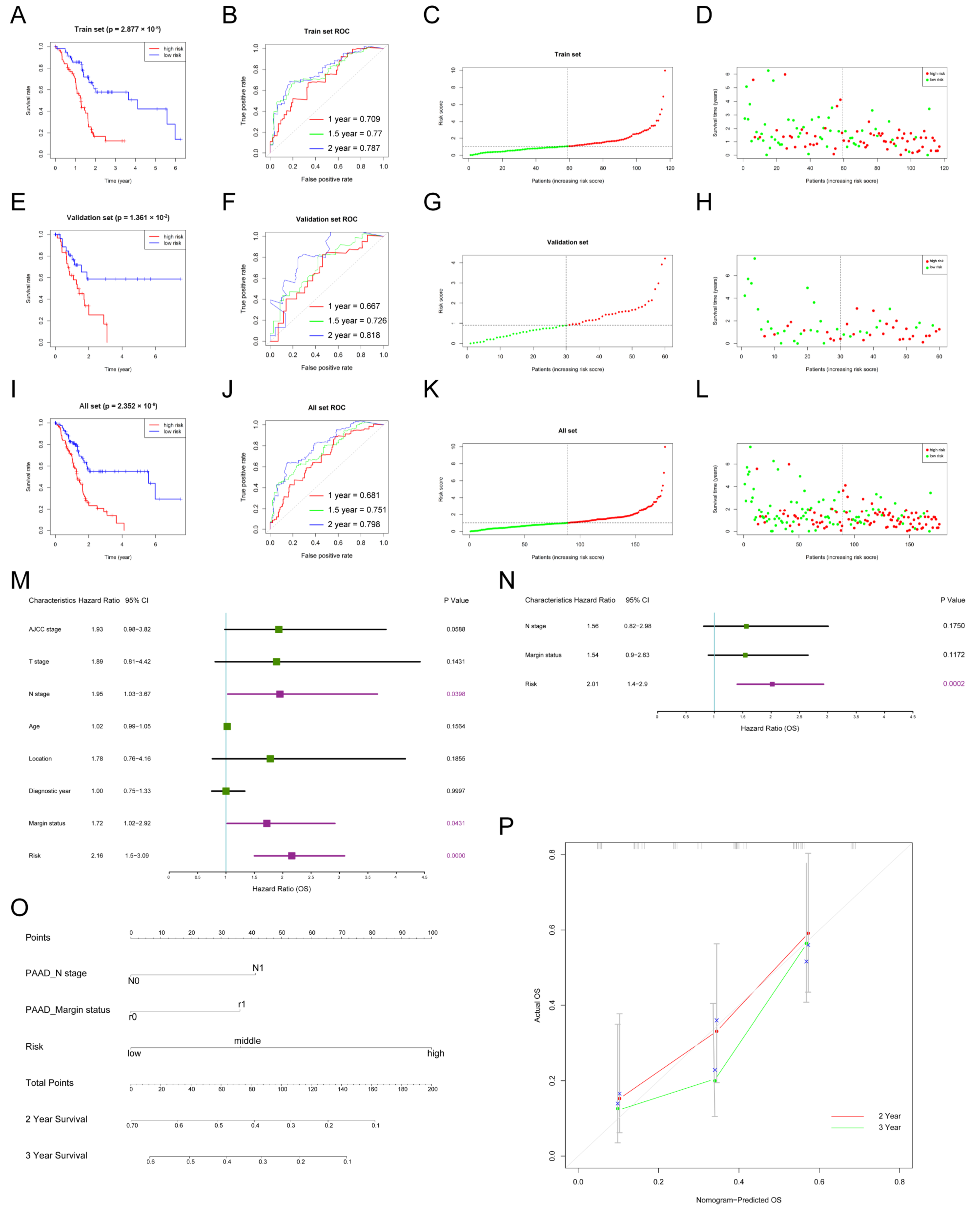
3.2. The Construction and Internal Validation of the Prognostic Model for PDAC
3.3. Nomogram to Predict the Survival of Patients with PDAC
3.4. The Prognostic Model Was Associated with Tumor Immune Infiltration Status
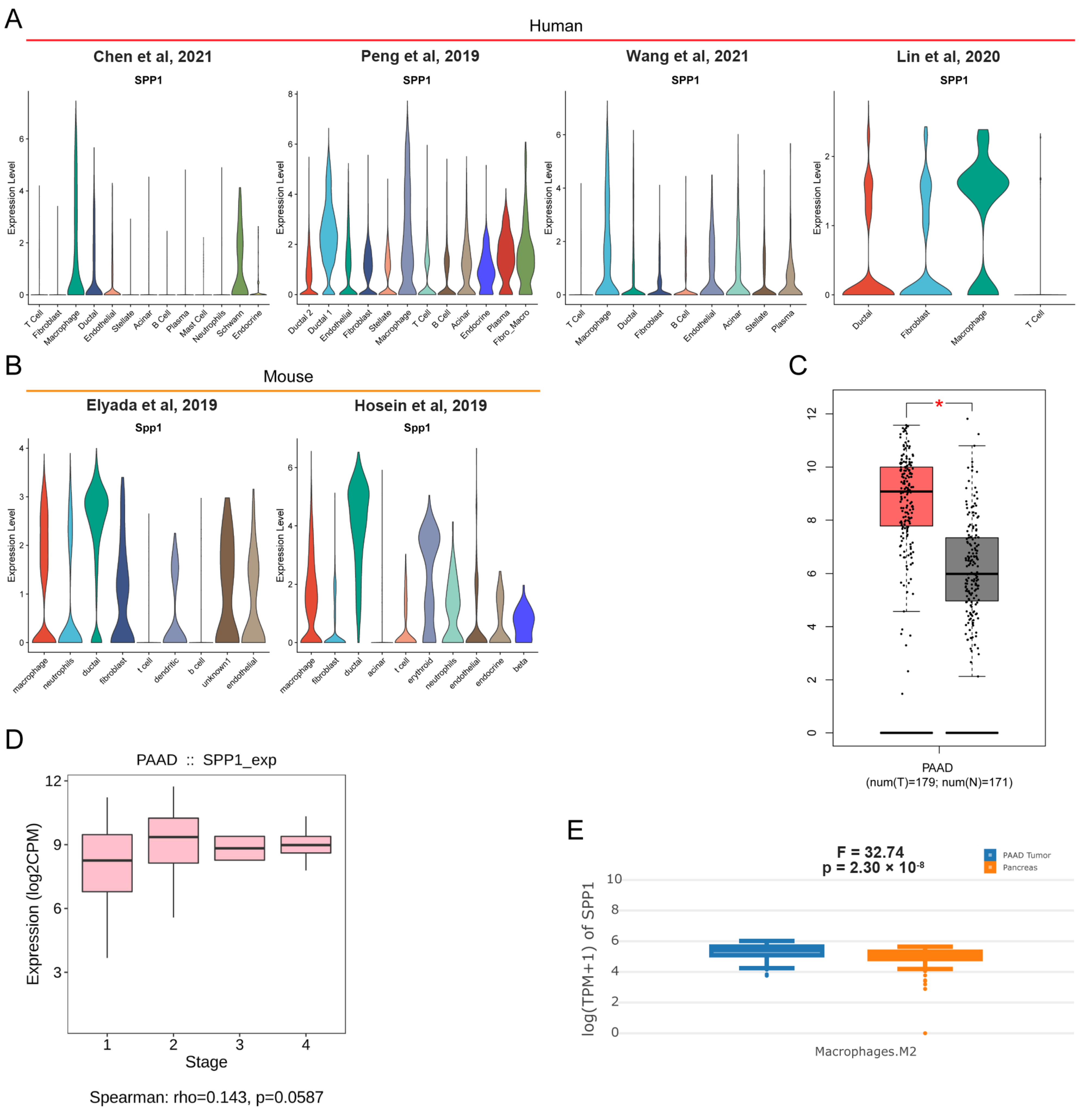
3.5. Pan-Cancer Landscape of SPP1 Was Delineated
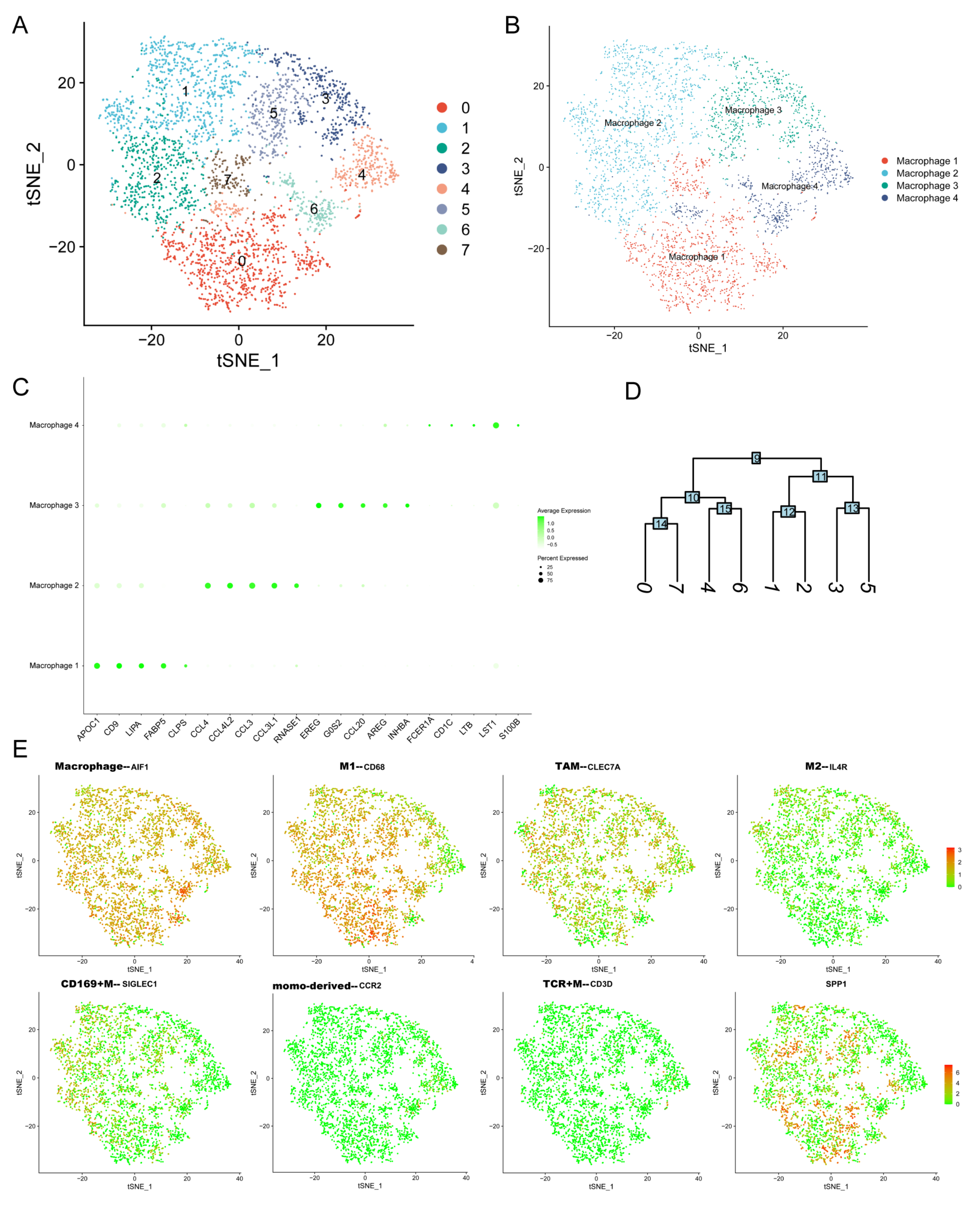
3.6. ScRNA-Seq Identified the Expression of SPP1 in Various Cell Subpopulations of PDAC
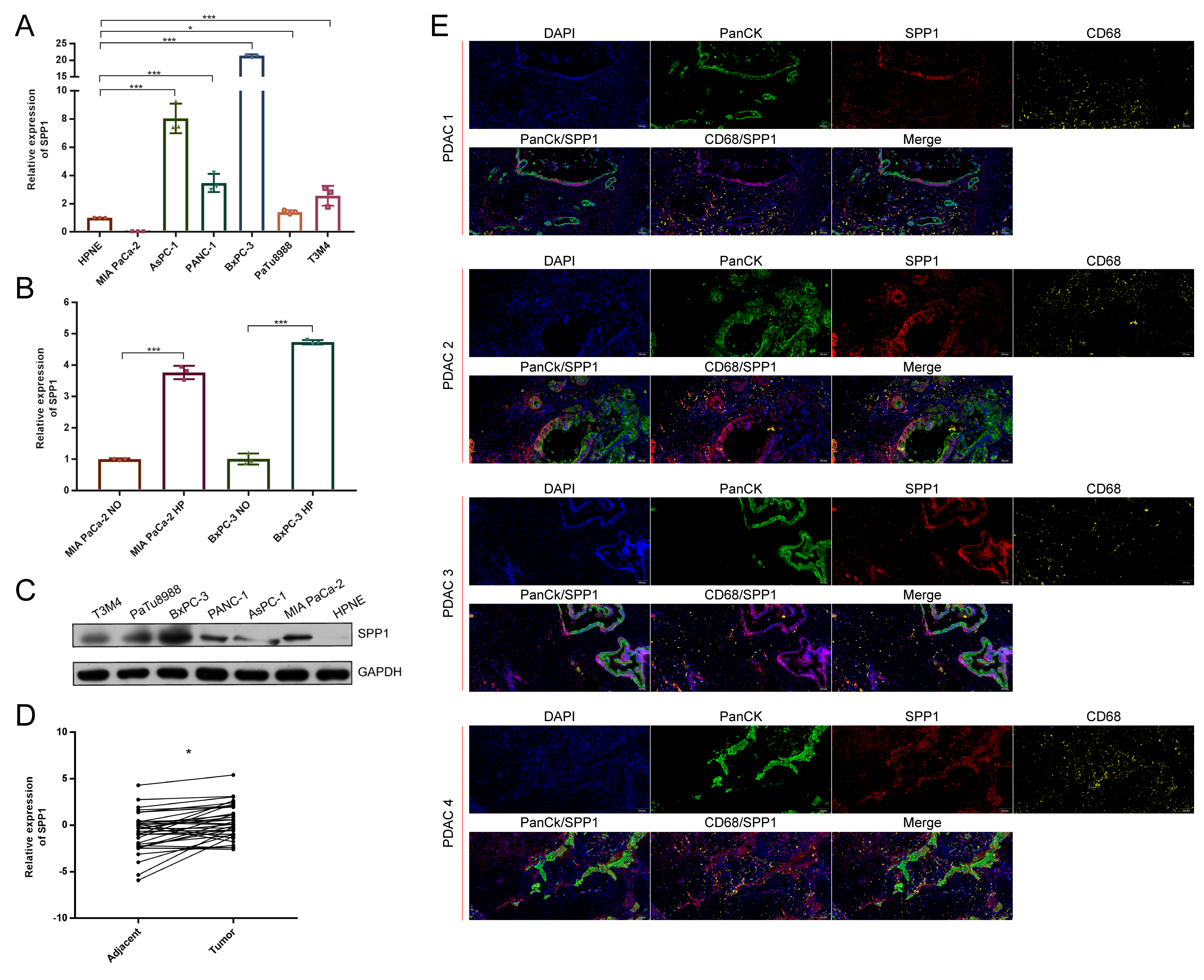
3.7. The Distribution of SPP1 in PDAC Was Verified by Multicolor IHC Stating
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Z.; Zou, W.B.; Wang, F.; Zhang, G.; Chen, K.; Hu, M.G.; Liu, R. Identification of the immune cell infiltration landscape in pancreatic cancer to assist immunotherapy. Future Oncol. 2021, 17, 4131–4143. [Google Scholar] [CrossRef]
- Velasco, R.M.; García, A.G.; Sánchez, P.J.; Sellart, I.M.; Sánchez-Arévalo Lobo, V.J. Tumour microenvironment and heterotypic interactions in pancreatic cancer. J. Physiol. Biochem. 2022, 1–14. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell. 2019, 75, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Noel, P.; Borazanci, E.H.; Lee, J.; Amini, A.; Han, I.W.; Heo, J.S.; Jameson, G.S.; Fraser, C.; Steinbach, M.; et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 2020, 12, 80. [Google Scholar] [CrossRef]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Sun, J.; Zhang, S.; Chen, J.; Wang, G.; Ju, S.; Wang, X. A novel methylation signature predicts inferior outcome of patients with PDAC. Aging (Albany NY) 2021, 13, 2851–2863. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, H.; Chen, Q.; Wu, W.; Chen, X.; Ran, L.; Si, G.; Tan, X. A Novel and Robust Long Noncoding RNA Panel to Predict the Prognosis of Pancreatic Cancer. DNA Cell Biol. 2020, 39, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Shao, Z.; Chen, S.; Wang, W.; Wang, Y.; Jiang, J.; Ma, Q.; Zhang, L. Construction of an RNA-Binding Protein-Related Prognostic Model for Pancreatic Adenocarcinoma Based on TCGA and GTEx Databases. Front. Genet. 2020, 11, 610350. [Google Scholar] [CrossRef]
- Yue, P.; Zhu, C.; Gao, Y.; Li, Y.; Wang, Q.; Zhang, K.; Gao, S.; Shi, Y.; Wu, Y.; Wang, B.; et al. Development of an autophagy-related signature in pancreatic adenocarcinoma. Biomed. Pharmacother. 2020, 126, 110080. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Z.; Yu, X.; Zhang, M.; Zheng, Q.; He, Y.; Guo, W. Immune Subtypes Based on Immune-Related lncRNA: Differential Prognostic Mechanism of Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 698296. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, X.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Development and validation of prognostic and diagnostic model for pancreatic ductal adenocarcinoma based on scRNA-seq and bulk-seq datasets. Hum. Mol. Genet. 2021, 31, 1705–1719. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Q.; Liu, X.; Wang, F.; Yang, Y.; Tian, X. Hypoxic pancreatic cancer derived exosomal miR-30b-5p promotes tumor angiogenesis by inhibitingGJA1 expression. Int. J. Biol. Sci. 2021, 18, 1220–1237. [Google Scholar]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Y.; Hou, Y.; Wang, Q.; Long, D.; Liu, X.; Tian, X.; Yang, Y. Single cell RNA-seq reveals the CCL5/SDC1 receptor-ligand interaction between T cells and tumor cells in pancreatic cancer. Cancer Lett. 2022, 545, 215834. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Rouzaut, A.; Motz, G.T.; Coukos, G. T-Cell and NK-Cell Infiltration into Solid Tumors: A Key Limiting Factor for Efficacious Cancer Immunotherapy. Cancer Discov. 2014, 4, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi Rad, H.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498. [Google Scholar] [CrossRef] [PubMed]
- Karpinsky, G.; Fatyga, A.; Krawczyk, M.A.; Chamera, M.; Sande, N.; Szmyd, D.; Izycka-Swieszewska, E.; Bien, E. Osteopontin: Its potential role in cancer of children and young adults. Biomark. Med. 2017, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Xu, B.H.; Zhou, D.L.; Ye, Z.L.; He, H.C.; Yang, X.H.; Zhang, X.; Liu, Q.; Ma, J.J.; Shao, Q.; et al. Polymorphisms in matricellular SPP1 and SPARC contribute to susceptibility to papillary thyroid cancer. Genomics 2020, 112, 4959–4967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, W.; Chen, Z.; Xiang, C. Upregulation of PD-L1 by SPP1 mediates macrophage polarization and facilitates immune escape in lung adenocarcinoma. Exp. Cell Res. 2017, 359, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hou, C.; Zheng, Z.; Lin, H.; Lv, G.; Zhou, D. Identification of Secreted Phosphoprotein 1 (SPP1) as a Prognostic Factor in Lower-Grade Gliomas. World Neurosurg. 2019, 130, e775–e785. [Google Scholar] [CrossRef]
- Li, S.; Yang, R.; Sun, X.; Miao, S.; Lu, T.; Wang, Y.; Wo, Y.; Jiao, W. Identification of SPP1 as a promising biomarker to predict clinical outcome of lung adenocarcinoma individuals. Gene 2018, 679, 398–404. [Google Scholar] [CrossRef]
- Zhang, W.G.; Fan, J.L.; Chen, Q.; Lei, C.P.; Qiao, B.; Liu, Q. SPP1 and AGER as potential prognostic biomarkers for lung adenocarcinoma. Oncol. Lett. 2018, 15, 7028–7036. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5, e129212. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Sasaki, S.I.; Baba, T. CCL4 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1231, 23–32. [Google Scholar]
- Ntanasis-Stathopoulos, I.; Fotiou, D.; Terpos, E. CCL3 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1231, 13–21. [Google Scholar] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 2021, 184, 404–421.e416. [Google Scholar] [CrossRef]
- Chen, D.T.; Davis-Yadley, A.H.; Huang, P.Y.; Husain, K.; Centeno, B.A.; Permuth-Wey, J.; Pimiento, J.M.; Malafa, M. Prognostic Fifteen-Gene Signature for Early Stage Pancreatic Ductal Adenocarcinoma. PLoS ONE 2015, 10, e0133562. [Google Scholar] [CrossRef]
- Ellsworth, K.A.; Eckloff, B.W.; Li, L.; Moon, I.; Fridley, B.L.; Jenkins, G.D.; Carlson, E.; Brisbin, A.; Abo, R.; Bamlet, W.; et al. Contribution of FKBP5 genetic variation to gemcitabine treatment and survival in pancreatic adenocarcinoma. PLoS ONE 2013, 8, e70216. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; He, P.; Wang, J.; Schetter, A.; Tang, W.; Funamizu, N.; Yanaga, K.; Uwagawa, T.; Satoskar, A.R.; Gaedcke, J.; et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Res. 2016, 76, 3838–3850. [Google Scholar] [CrossRef]
- Liu, W.; Lu, Y.; Zhang, D.; Shi, L.; Zu, G.; Yan, H.; Sun, D. MicroRNA-708 inhibits the proliferation and chemoresistance of pancreatic cancer cells. Biocell 2020, 44, 73–80. [Google Scholar] [CrossRef]
- Sin-Chan, P.; Li, B.K.; Ho, B.; Fonseca, A.; Huang, A. Molecular Classification and Management of Rare Pediatric Embryonal Brain Tumors. Curr. Oncol. Rep. 2018, 20, 69. [Google Scholar] [CrossRef]
- Timar, J. Molecular classification of pancreatic cancer. Magy Onkol. 2021, 65, 201–205. [Google Scholar] [PubMed]
- Barry, S.T.; Ludbrook, S.B.; Murrison, E.; Horgan, C.M. Analysis of the alpha4beta1 integrin-osteopontin interaction. Exp. Cell Res. 2000, 258, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, Y.U.; Sleeman, J.; Fujii, H.; Herrlich, P.; Hotta, H.; Tanaka, K.; Chikuma, S.; Yagita, H.; Okumura, K.; Murakami, M.; et al. CD44 variants but not CD44s cooperate with beta 1-containing integrins to permit cells to bind to osteopontin independently of arginine-glycine-aspartic acid, thereby stimulating cell motility and chemotaxis. Cancer Res. 1999, 59, 219–226. [Google Scholar] [PubMed]
- Deng, G.; Zeng, F.; Su, J.; Zhao, S.; Hu, R.; Zhu, W.; Hu, S.; Chen, X.; Yin, M. BET inhibitor suppresses melanoma progression via the noncanonical NF-kappa B/SPP1 pathway. Theranostics 2020, 10, 11428–11443. [Google Scholar] [CrossRef]
- Chen, X.; Xiong, D.; Ye, L.; Yang, H.; Mei, S.; Wu, J.; Chen, S.; Mi, R. SPP1 inhibition improves the cisplatin chemo-sensitivity of cervical cancer cell lines. Cancer Chemother. Pharmacol. 2019, 83, 603–613. [Google Scholar] [CrossRef]
- Assidi, M.; Gomaa, W.; Jafri, M.; Hanbazazh, M.; Al-Ahwal, M.; Pushparaj, P.; Al-Harbi, A.; Al-Qahtani, M.; Buhmeida, A.; Al-Maghrabi, J. Prognostic value of Osteopontin (SPP1) in colorectal carcinoma requires a personalized molecular approach. Tumour Biol. 2019, 41, 1010428319863627. [Google Scholar] [CrossRef]
- Tang, H.P.; Chen, J.Y.; Han, X.L.; Feng, Y.; Wang, F. Upregulation of SPP1 Is a Marker for Poor Lung Cancer Prognosis and Contributes to Cancer Progression and Cisplatin Resistance. Front. Cell Dev. Biol. 2021, 9, 1109. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.; Wang, Q.; Liu, X.; Wang, F.; Ma, Y.; Zhang, S.; Shao, Z.; Yang, Y.; Tian, X. Single Cell RNA-Seq Identifies Immune-Related Prognostic Model and Key Signature-SPP1 in Pancreatic Ductal Adenocarcinoma. Genes 2022, 13, 1760. https://doi.org/10.3390/genes13101760
Chen K, Wang Q, Liu X, Wang F, Ma Y, Zhang S, Shao Z, Yang Y, Tian X. Single Cell RNA-Seq Identifies Immune-Related Prognostic Model and Key Signature-SPP1 in Pancreatic Ductal Adenocarcinoma. Genes. 2022; 13(10):1760. https://doi.org/10.3390/genes13101760
Chicago/Turabian StyleChen, Kai, Qi Wang, Xinxin Liu, Feng Wang, Yongsu Ma, Shupeng Zhang, Zhijiang Shao, Yinmo Yang, and Xiaodong Tian. 2022. "Single Cell RNA-Seq Identifies Immune-Related Prognostic Model and Key Signature-SPP1 in Pancreatic Ductal Adenocarcinoma" Genes 13, no. 10: 1760. https://doi.org/10.3390/genes13101760
APA StyleChen, K., Wang, Q., Liu, X., Wang, F., Ma, Y., Zhang, S., Shao, Z., Yang, Y., & Tian, X. (2022). Single Cell RNA-Seq Identifies Immune-Related Prognostic Model and Key Signature-SPP1 in Pancreatic Ductal Adenocarcinoma. Genes, 13(10), 1760. https://doi.org/10.3390/genes13101760