

Using Sequence Similarity Based on CKSNP Features and a Graph Neural Network Model to Identify miRNA–Disease Associations


Abstract
1. Introduction
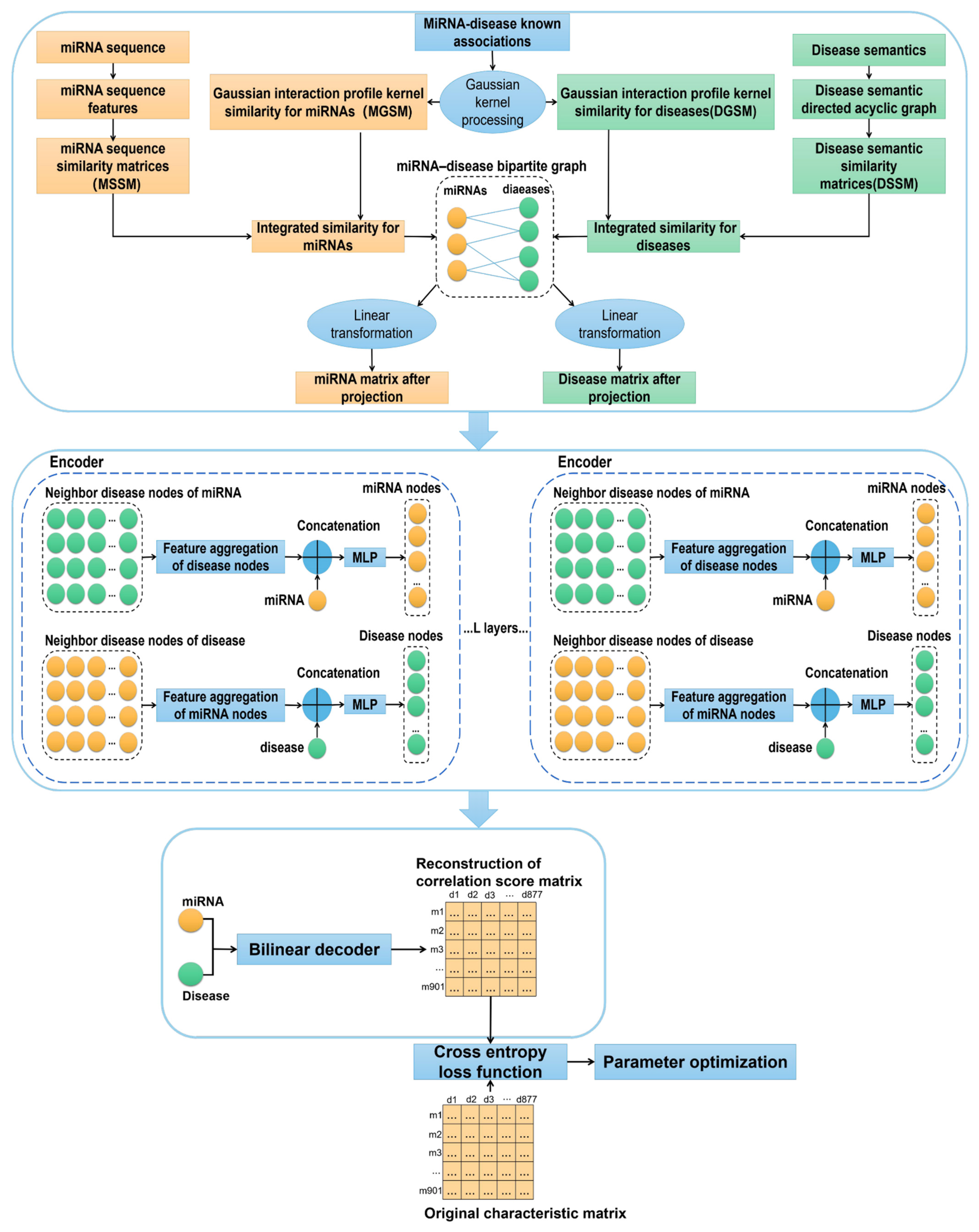
2. Materials and Methods
2.1. Human miRNA–Disease Associations
2.2. miRNA Sequence Similarity
2.3. Disease Semantic Similarity
2.4. Gaussian Interaction Profile Kernel Similarity for miRNAs and Diseases
2.5. Integrated Similarity for miRNAs and Diseases
2.6. Graph Auto-Encoder
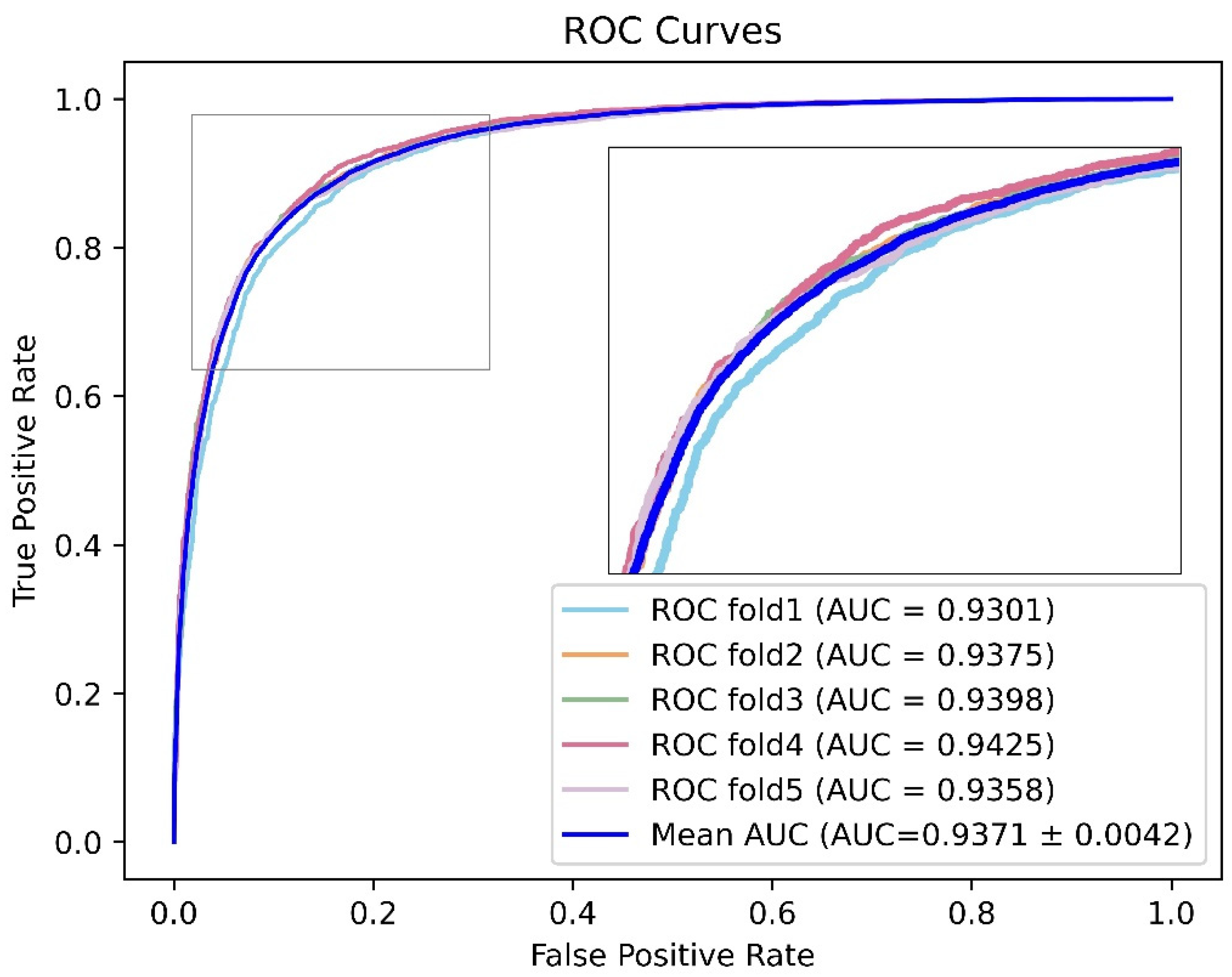
2.7. Model Evaluation
3. Results
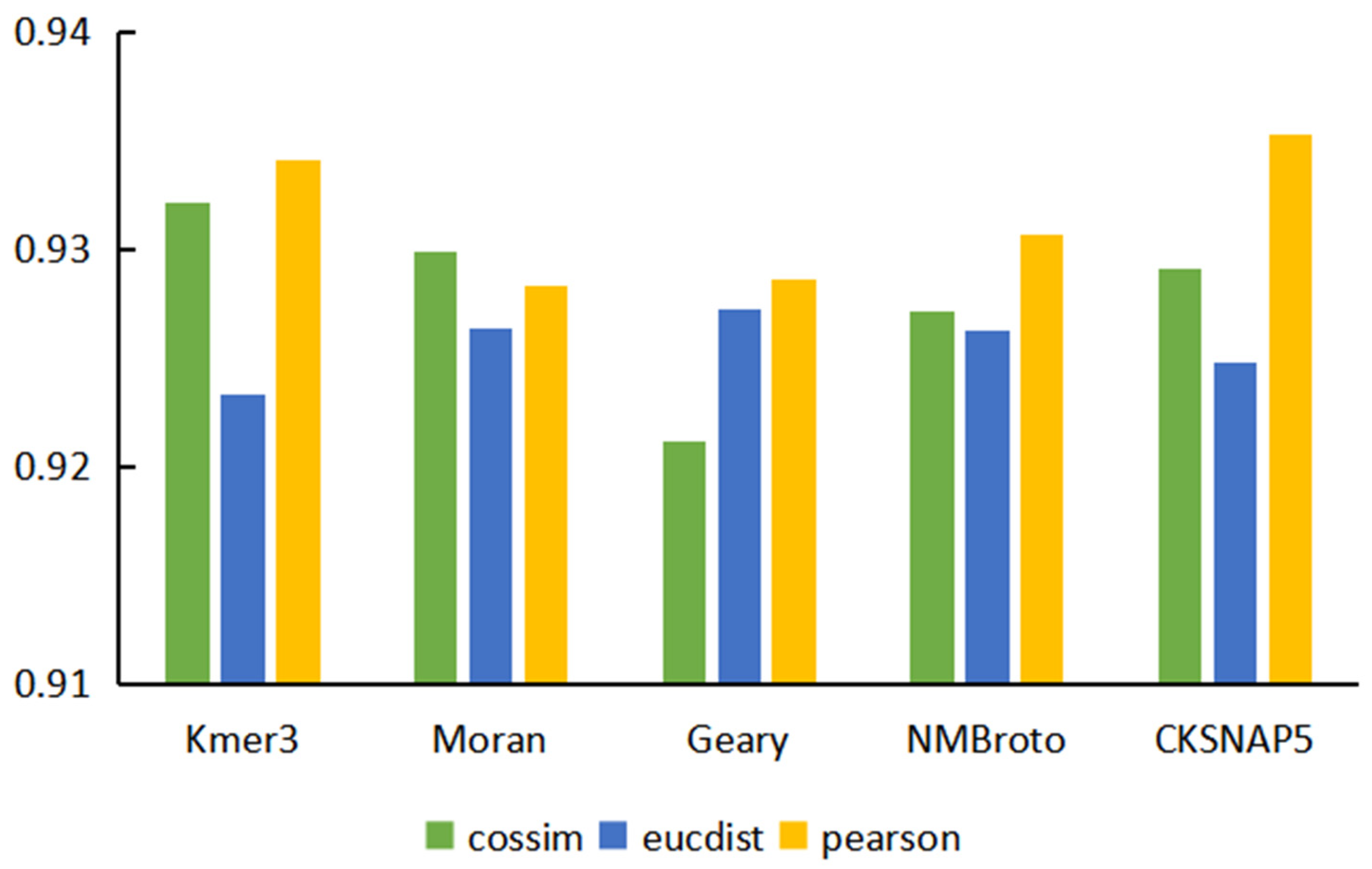
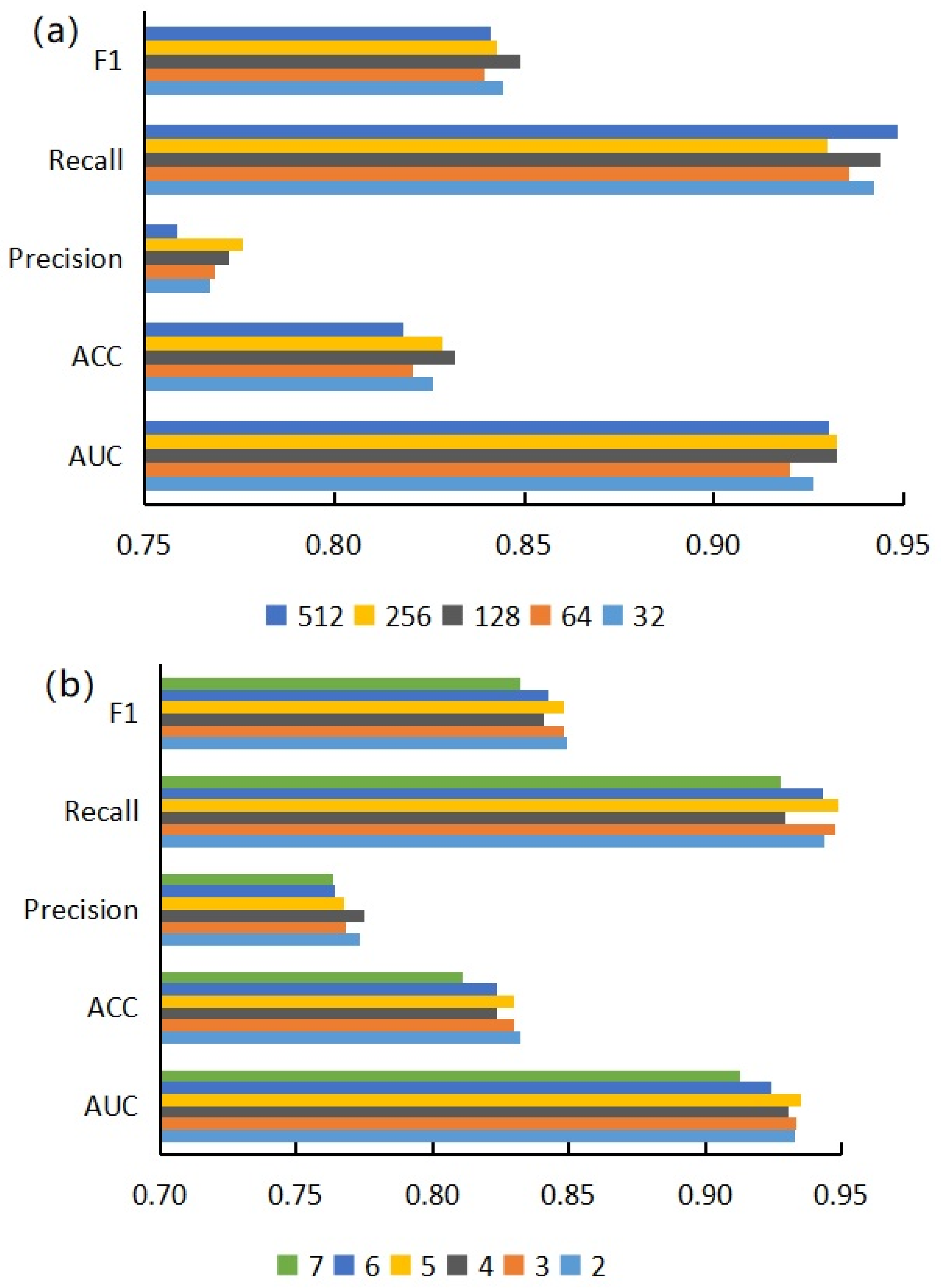
3.1. Performance Evaluation of Graph Neural Network Prediction Model Based on Single Features
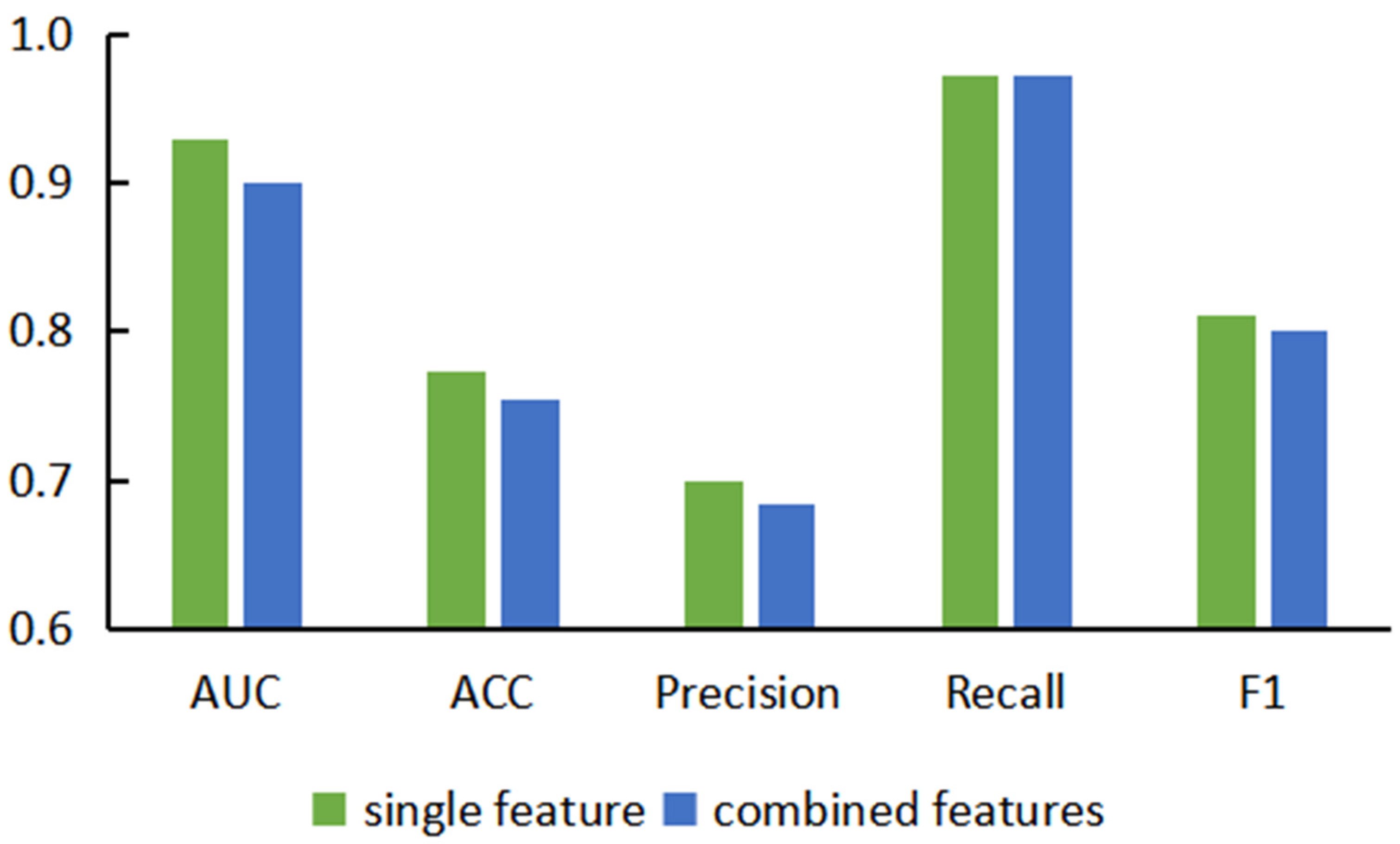
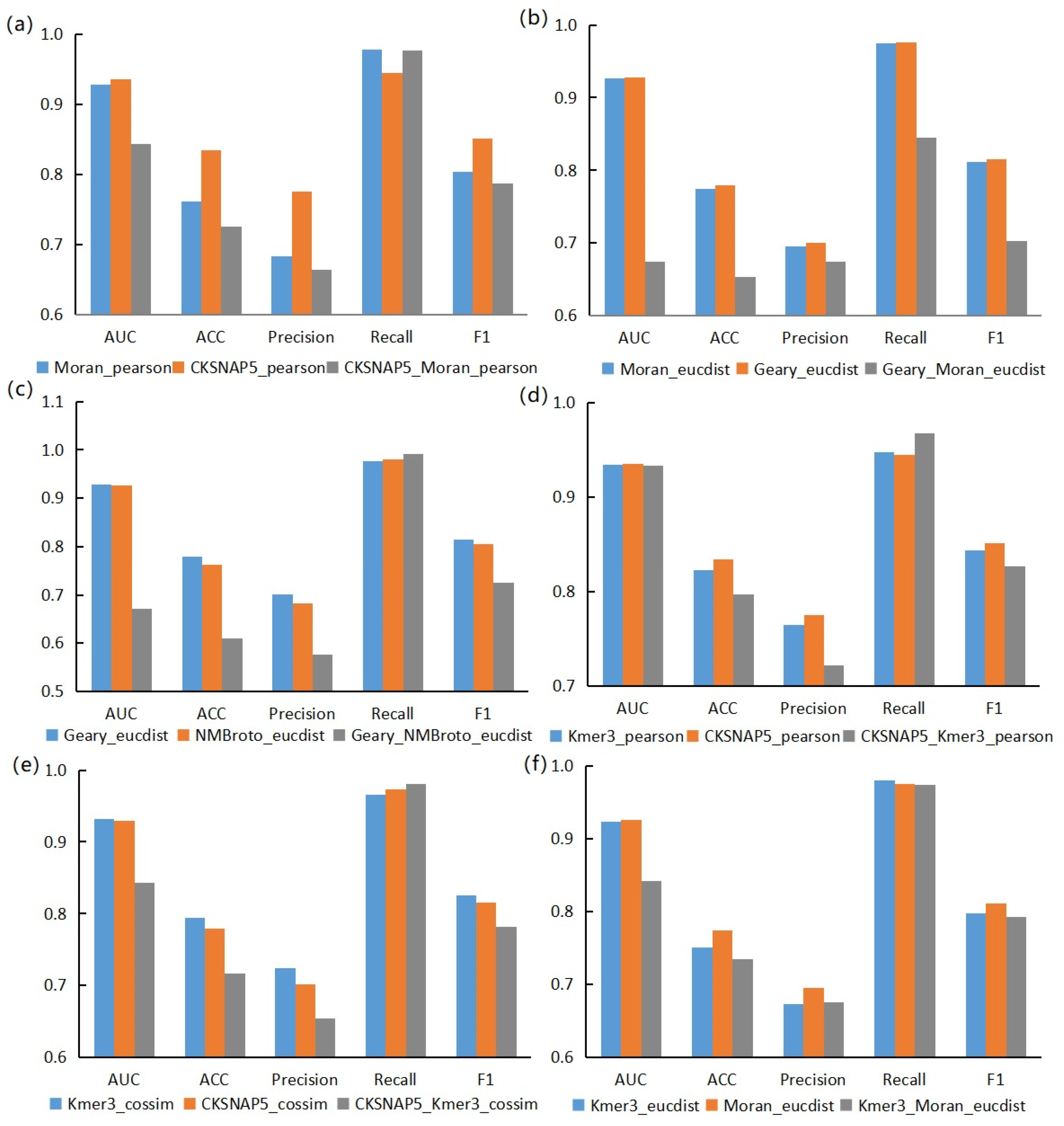
3.2. Performance Evaluation of Graph Neural Network Prediction Model Based on Combined Features
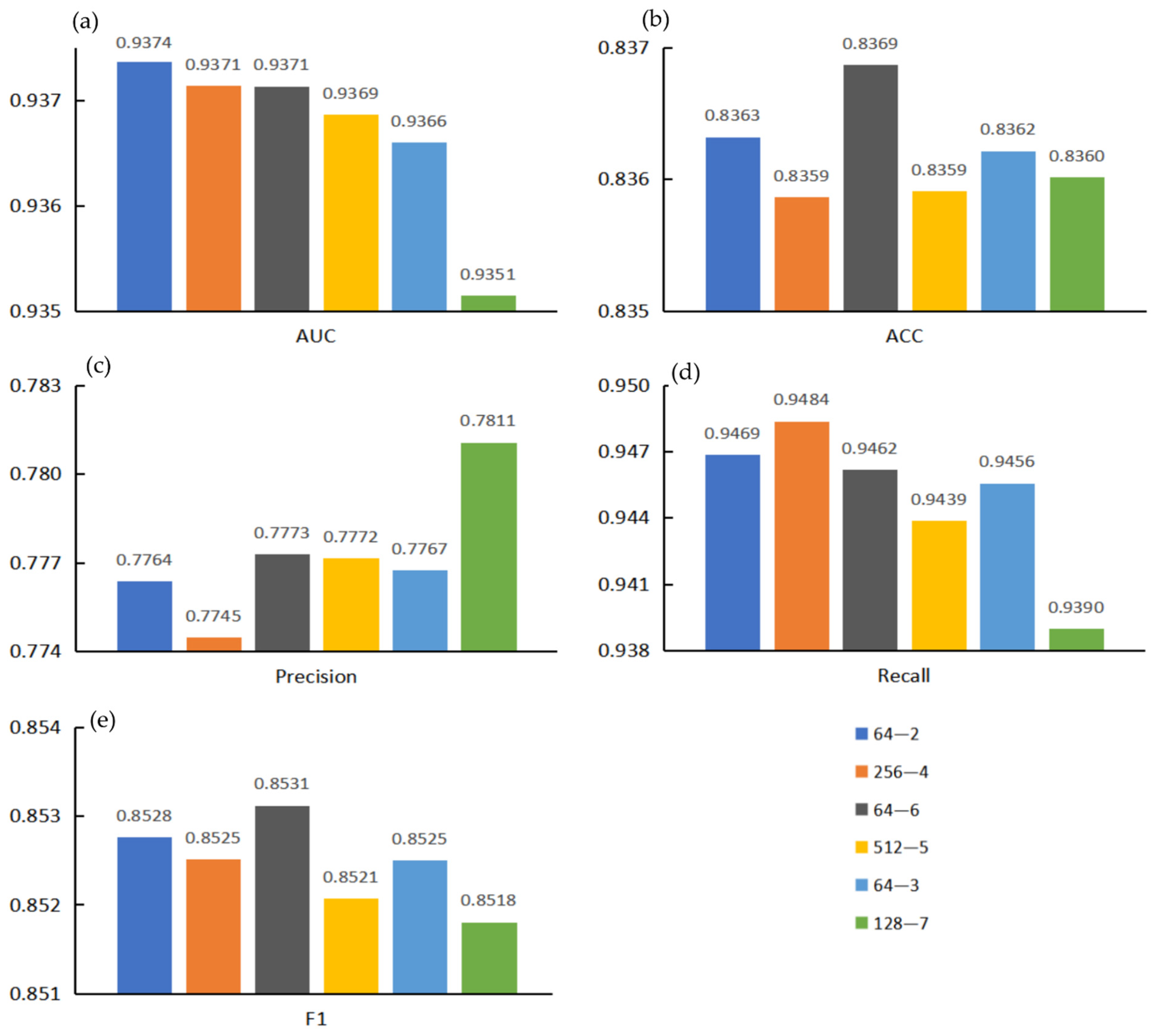
3.3. Effects of Projection Dimension and Encoder Layers on Model Performance
3.4. Performance Evaluation and Comparative Analysis of Related Models
3.5. Case Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crick, F.H.C.; Barnett, L.; Brenner, S.; Watts-Tobin, R.J. General Nature of the Genetic Code for Proteins. Nature 1961, 192, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Yanofsky, C. Establishing the Triplet Nature of the Genetic Code. Cell 2007, 128, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Bhattacharya, S.; Rawat, B.S.; Kumar, A.; Kumar, A.; Niraj, K.; Chande, A.; Gandhi, P.; Khetan, D.; Aggarwal, A. MicroRNA-30e-5p has an integrated role in the regulation of the innate immune response during virus infection and systemic lupus erythematosus. iScience 2020, 23, 101322. [Google Scholar] [CrossRef]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor Origin Detection with Tissue-Specific miRNA and DNA methylation Markers. Bioinformatics 2018, 34, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; You, Z.-H.; Guo, Z.-H.; Yi, H.-C.; Chen, Z.-H.; Cao, M.-Y. MIPDH: A Novel Computational Model for Predicting microRNA–mRNA Interactions by DeepWalk on a Heterogeneous Network. ACS Omega 2020, 5, 17022–17032. [Google Scholar] [CrossRef]
- Freeman, W.; Walker, S.; Vrana, K. Quantitative RT-PCR: Pitfalls and potential. BioTechniques 1999, 26, 112–122; 124–125. [Google Scholar] [CrossRef]
- Várallyay, É.; Burgyán, J.; Havelda, Z. MicroRNA detection by northern blotting using locked nucleic acid probes. Nat. Protoc. 2008, 3, 190–196. [Google Scholar] [CrossRef]
- Baskerville, S.; Bartel, D.P. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. Rna 2005, 11, 241–247. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, X.; Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 2016, 17, 193–203. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Zhao, Q.; You, Z.H. MicroRNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2019, 20, 515–539. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Hao, Y.; Wang, G.; Juan, L.; Zhang, T.; Teng, M.; Liu, Y.; Wang, Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 2010, 4 (Suppl. S1), S2. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zeng, X.; He, Z.; Zou, Q. Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE ACM Trans. Comput. Biol. Bioinform. 2017, 14, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Liu, L.; Lu, L.; Zou, Q. Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 2018, 34, 2425–2432. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Z.; Guo, W.; Yang, W.; Huang, F. A fast linear neighborhood similarity-based network link inference method to predict microRNA-disease associations. IEEE ACM Trans. Comput. Biol. Bioinform. 2019, 18, 405–415. [Google Scholar] [CrossRef]
- Mørk, S.; Pletscher-Frankild, S.; Palleja Caro, A.; Gorodkin, J.; Jensen, L.J. Protein-driven inference of miRNA–disease associations. Bioinformatics 2013, 30, 392–397. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef]
- Chen, X.; Wang, C.C.; Yin, J.; You, Z.H. Novel Human miRNA-Disease Association Inference Based on Random Forest. Mol. Ther. Nucleic Acids 2018, 13, 568–579. [Google Scholar] [CrossRef]
- Zeng, X.; Wang, W.; Deng, G.; Bing, J.; Zou, Q. Prediction of potential disease-associated microRNAs by using neural network. Mol. Ther. Nucleic Acids 2019, 16, 566–575. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, S.; Wu, Q.; Azim, R.; Li, W. Predicting potential miRNA-disease associations by combining gradient boosting decision tree with logistic regression. Comput. Biol. Chem. 2020, 85, 107200. [Google Scholar] [CrossRef]
- Ma, Y.; He, T.; Ge, L.; Zhang, C.; Jiang, X. MiRNA-disease interaction prediction based on kernel neighborhood similarity and multi-network bidirectional propagation. BMC Med. Genom. 2019, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.; Ke, H.; Guo, M.; Guo, Y.; Li, J.; Jian, D.; Yong, L.; Dai, Q.; Jin, L.; Teng, Z. Correction: Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS ONE 2013, 8, 3752034. [Google Scholar] [CrossRef]
- Lu, X.; Gao, Y.; Zhu, Z.; Ding, L.; Wang, X.; Liu, F.; Li, J. A Constrained Probabilistic Matrix Decomposition Method for Predicting miRNA-disease Associations. Curr. Bioinform. 2021, 16, 524–533. [Google Scholar] [CrossRef]
- Tian, L.; Wang, S.-L. Exploring miRNA Sponge Networks of Breast Cancer by Combining miRNA-disease-lncRNA and miRNA-target Networks. Curr. Bioinform. 2021, 16, 385–394. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, Q.; Liang, C. Prediction of lncRNA-disease Associations Based on Robust Multi-label Learning. Curr. Bioinform. 2021, 16, 1179–1189. [Google Scholar] [CrossRef]
- Zhang, Y.; Duan, G.; Yan, C.; Yi, H.; Wu, F.-X.; Wang, J. MDAPlatform: A Component-based Platform for Constructing and Assessing miRNA-disease Association Prediction Methods. Curr. Bioinform. 2021, 16, 710–721. [Google Scholar] [CrossRef]
- Zhu, Q.; Fan, Y.; Pan, X. Fusing Multiple Biological Networks to Effectively Predict miRNA-disease Associations. Curr. Bioinform. 2021, 16, 371–384. [Google Scholar] [CrossRef]
- Jiang, L.; Zhu, J. Review of MiRNA-disease association prediction. Curr. Protein Pept. Sci. 2020, 21, 1044–1053. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and between score for MiRNA-disease association prediction. Sci. Rep. 2016, 6, 21106. [Google Scholar] [CrossRef]
- Yao, D.; Zhan, X.; Kwoh, C.-K. An improved random forest-based computational model for predicting novel miRNA-disease associations. BMC Bioinform. 2019, 20, 624. [Google Scholar] [CrossRef]
- Ji, B.-Y.; You, Z.-H.; Cheng, L.; Zhou, J.-R.; Alghazzawi, D.; Li, L.-P. Predicting miRNA-disease association from heterogeneous information network with GraRep embedding model. Sci. Rep. 2020, 10, 6658. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.-Y.; You, Z.-H.; Wang, Y.; Li, Z.-W.; Wong, L. DANE-MDA: Predicting microRNA-disease associations via deep attributed network embedding. iScience 2021, 24, 102455. [Google Scholar] [CrossRef]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y.; et al. dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA–disease associations. Nucleic Acids Res. 2018, 47, D1013–D1017. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Clarence Yan, C.; Luo, C.; Ji, W.; Zhang, Y.; Dai, Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci. Rep. 2015, 5, 11338. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Huang, Z.-A.; Zhu, Z.; Yan, G.-Y.; Li, Z.-W.; Wen, Z.; Chen, X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, 1005455. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, H.; Lyu, C.; Liang, C. LLCMDA: A Novel Method for Predicting miRNA Gene and Disease Relationship Based on Locality-Constrained Linear Coding. Front. Genet. 2018, 9, 576. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhu, C.-C.; Yin, J. Ensemble of decision tree reveals potential miRNA-disease associations. PLoS Comput. Biol. 2019, 15, 1007209. [Google Scholar] [CrossRef]
- Yu, S.P.; Liang, C.; Xiao, Q.; Li, G.H.; Ding, P.J.; Luo, J.W. MCLPMDA: A novel method for mi RNA-disease association prediction based on matrix completion and label propagation. J. Cell. Mol. Med. 2019, 23, 1427–1438. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Nie, R.; You, Z.-H.; Bao, W. A graph auto-encoder model for miRNA-disease associations prediction. Brief. Bioinform. 2020, 22, bbaa240. [Google Scholar] [CrossRef]
- Birks, D.K.; Barton, V.N.; Donson, A.M.; Handler, M.H.; Vibhakar, R.; Foreman, N.K. Survey of MicroRNA expression in pediatric brain tumors. Pediatr. Blood Cancer 2011, 56, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Alder, H.; Taccioli, C.; Chen, H.; Jiang, Y.; Smalley, K.J.; Fadda, P.; Ozer, H.G.; Huebner, K.; Farber, J.L.; Croce, C.M.; et al. Dysregulation of miR-31 and miR-21 induced by zinc deficiency promotes esophageal cancer. Carcinogenesis 2012, 33, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar] [CrossRef]
- Linehan, W.M. Genetic basis of kidney cancer: Role of genomics for the development of disease-based therapeutics. Genome Res. 2012, 22, 2089–2100. [Google Scholar] [CrossRef]
- Senanayake, U.; Das, S.; Vesely, P.; Alzoughbi, W.; Fröhlich, L.F.; Chowdhury, P.; Leuschner, I.; Hoefler, G.; Guertl, B. miR-192, miR-194, miR-215, miR-200c and miR-141 are downregulated and their common target ACVR2B is strongly expressed in renal childhood neoplasms. Carcinogenesis 2012, 33, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.S.; Shahryari, V.; Deng, G.; Thamminana, S.; Saini, S.; Majid, S.; Chang, I.; Hirata, H.; Ueno, K.; Yamamura, S. Correction: Up-Regulation of MicroRNA-21 Correlates with Lower Kidney Cancer Survival. PLoS ONE 2012, 7, 31060. [Google Scholar] [CrossRef]
- Kim, K.; Taylor, S.L.; Ganti, S.; Guo, L.; Weiss, R.H. Urine Metabolomic Analysis Identifies Potential Biomarkers and Pathogenic Pathways in Kidney Cancer. Omics A J. Integr. Biol. 2011, 15, 293–303. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | AUC (%) |
|---|---|
| PBMDA | 91.72 |
| LLCMDA | 91.90 |
| EDTMDA | 91.92 |
| GBDTLR | 92.74 |
| MCLPMDA | 93.20 |
| GAEMDA | 93.56 |
| Our Model | 93.71 |
| miRNA | dbDEMC | miRNA | dbDEMC |
|---|---|---|---|
| hsa-mir-586 | Confirmed | hsa-mir-329-5p | Confirmed |
| hsa-mir-208b-5p | Confirmed | hsa-mir-1264 | Confirmed |
| hsa-mir-376b-5p | Confirmed | hsa-mir-618 | Confirmed |
| hsa-mir-3613-5p | Confirmed | hsa-mir-599 | Confirmed |
| hsa-mir-4775 | Confirmed | hsa-mir-517c-3p | Unconfirmed |
| hsa-mir-544a | Confirmed | hsa-mir-384 | Confirmed |
| hsa-mir-450a-5p | Confirmed | hsa-mir-581 | Confirmed |
| hsa-mir-376c-5p | Confirmed | hsa-mir-578 | Confirmed |
| hsa-mir-376a-5p | Confirmed | hsa-mir-19b-2-5p | Confirmed |
| hsa-mir-190a-5p | Confirmed | hsa-mir-552-5p | Confirmed |
| hsa-mir-875-5p | Confirmed | hsa-mir-5590-5p | Confirmed |
| hsa-mir-3682-5p | Confirmed | hsa-mir-450a-1-3p | Confirmed |
| hsa-mir-302f | Confirmed | hsa-mir-454-5p | Confirmed |
| hsa-mir-5586-5p | Confirmed | hsa-mir-942-5p | Confirmed |
| hsa-mir-450b-5p | Confirmed | hsa-mir-548l | Confirmed |
| hsa-mir-576-5p | Confirmed | hsa-mir-548k | Confirmed |
| hsa-mir-4295 | Confirmed | hsa-mir-1185-5p | Confirmed |
| hsa-mir-1282 | Confirmed | hsa-mir-548am-5p | Confirmed |
| hsa-mir-5009-5p | Confirmed | hsa-mir-613 | Confirmed |
| hsa-mir-655-5p | Confirmed | hsa-mir-1248 | Confirmed |
| hsa-mir-16-2-3p | Confirmed | hsa-mir-544b | Confirmed |
| hsa-mir-548d-5p | Confirmed | hsa-mir-3913-5p | Confirmed |
| hsa-mir-1179 | Confirmed | hsa-mir-548c-5p | Confirmed |
| hsa-mir-876-5p | Confirmed | hsa-mir-570-5p | Unconfirmed |
| hsa-mir-1206 | Unconfirmed | hsa-mir-651-5p | Confirmed |
| miRNA | dbDEMC | miRNA | dbDEMC |
|---|---|---|---|
| hsa-mir-1179 | Confirmed | hsa-mir-450b-5p | Confirmed |
| hsa-mir-1206 | Confirmed | hsa-mir-4775 | Confirmed |
| hsa-mir-1264 | Confirmed | hsa-mir-493-5p | Confirmed |
| hsa-mir-1282 | Confirmed | hsa-mir-495-5p | Confirmed |
| hsa-mir-135a-5p | Confirmed | hsa-mir-5009-5p | Confirmed |
| hsa-mir-136-5p | Confirmed | hsa-mir-517c-3p | Confirmed |
| hsa-mir-16-2-3p | Confirmed | hsa-mir-544a | Confirmed |
| hsa-mir-190a-5p | Confirmed | hsa-mir-545-5p | Confirmed |
| hsa-mir-196a-5p | Confirmed | hsa-mir-548d-5p | Confirmed |
| hsa-mir-199b-5p | Confirmed | hsa-mir-552-5p | Unconfirmed |
| hsa-mir-19b-2-5p | Confirmed | hsa-mir-5586-5p | Confirmed |
| hsa-mir-202-5p | Confirmed | hsa-mir-5590-5p | Confirmed |
| hsa-mir-208b-5p | Confirmed | hsa-mir-576-5p | Confirmed |
| hsa-mir-29a-5p | Confirmed | hsa-mir-578 | Confirmed |
| hsa-mir-329-5p | Unconfirmed | hsa-mir-581 | Confirmed |
| hsa-mir-3613-5p | Confirmed | hsa-mir-586 | Confirmed |
| hsa-mir-3682-5p | Confirmed | hsa-mir-599 | Confirmed |
| hsa-mir-376a-2-5p | Confirmed | hsa-mir-618 | Confirmed |
| hsa-mir-376a-5p | Confirmed | hsa-mir-655-5p | Confirmed |
| hsa-mir-376c-5p | Confirmed | hsa-mir-7-5p | Confirmed |
| hsa-mir-384 | Confirmed | hsa-mir-875-5p | Confirmed |
| hsa-mir-4295 | Confirmed | hsa-mir-876-5p | Confirmed |
| hsa-mir-4423-5p | Confirmed | hsa-mir-95-5p | Confirmed |
| hsa-mir-450a-1-3p | Unconfirmed | hsa-mir-9-5p | Confirmed |
| hsa-mir-450a-5p | Confirmed | hsa-mir-29b-1-5p | Confirmed |
| miRNA | dbDEMC | miRNA | dbDEMC |
|---|---|---|---|
| hsa-mir-105-5p | Confirmed | hsa-mir-449a | Confirmed |
| hsa-mir-1179 | Confirmed | hsa-mir-449c-5p | Confirmed |
| hsa-mir-1204 | Confirmed | hsa-mir-4775 | Confirmed |
| hsa-mir-1244 | Confirmed | hsa-mir-4795-5p | Unconfirmed |
| hsa-mir-1264 | Confirmed | hsa-mir-517c-3p | Confirmed |
| hsa-mir-1267 | Confirmed | hsa-mir-5193 | Unconfirmed |
| hsa-mir-1282 | Confirmed | hsa-mir-520h | Unconfirmed |
| hsa-mir-1284 | Confirmed | hsa-mir-543 | Confirmed |
| hsa-mir-1322 | Confirmed | hsa-mir-548c-5p | Unconfirmed |
| hsa-mir-135b-5p | Confirmed | hsa-mir-5692b | Unconfirmed |
| hsa-mir-136-5p | Confirmed | hsa-mir-576-5p | Confirmed |
| hsa-mir-147b-5p | Unconfirmed | hsa-mir-577 | Confirmed |
| hsa-mir-149-5p | Confirmed | hsa-mir-586 | Confirmed |
| hsa-mir-18b-5p | Confirmed | hsa-mir-606 | Confirmed |
| hsa-mir-202-5p | Confirmed | hsa-mir-616-5p | Confirmed |
| hsa-mir-212-5p | Confirmed | hsa-mir-626 | Unconfirmed |
| hsa-mir-23c | Confirmed | hsa-mir-633 | Confirmed |
| hsa-mir-3120-5p | Unconfirmed | hsa-mir-644a | Unconfirmed |
| hsa-mir-3149 | Confirmed | hsa-mir-645 | Confirmed |
| hsa-mir-32-5p | Unconfirmed | hsa-mir-764 | Unconfirmed |
| hsa-mir-340-5p | Confirmed | hsa-mir-889-5p | Unconfirmed |
| hsa-mir-3662 | Confirmed | hsa-mir-934 | Confirmed |
| hsa-mir-3682-5p | Unconfirmed | hsa-mir-942-5p | Confirmed |
| hsa-mir-4295 | Confirmed | hsa-mir-943 | Confirmed |
| hsa-mir-4443 | Confirmed | hsa-mir-944 | Confirmed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Fan, Y.; Zhang, Y.; Lv, Z. Using Sequence Similarity Based on CKSNP Features and a Graph Neural Network Model to Identify miRNA–Disease Associations. Genes 2022, 13, 1759. https://doi.org/10.3390/genes13101759
Li M, Fan Y, Zhang Y, Lv Z. Using Sequence Similarity Based on CKSNP Features and a Graph Neural Network Model to Identify miRNA–Disease Associations. Genes. 2022; 13(10):1759. https://doi.org/10.3390/genes13101759
Chicago/Turabian StyleLi, Mingxin, Yu Fan, Yiting Zhang, and Zhibin Lv. 2022. "Using Sequence Similarity Based on CKSNP Features and a Graph Neural Network Model to Identify miRNA–Disease Associations" Genes 13, no. 10: 1759. https://doi.org/10.3390/genes13101759
APA StyleLi, M., Fan, Y., Zhang, Y., & Lv, Z. (2022). Using Sequence Similarity Based on CKSNP Features and a Graph Neural Network Model to Identify miRNA–Disease Associations. Genes, 13(10), 1759. https://doi.org/10.3390/genes13101759