
De Novo Assembly Transcriptome Analysis Reveals the Preliminary Molecular Mechanism of Primordium Formation in Pleurotus tuoliensis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. P. tuoliensis Cultivation
2.2. RNA Extraction
2.3. Library Preparation and RNA-seq
2.4. Transcriptome Assembly and Gene Function Annotation
2.5. Analysis of Differentially Expressed Genes (DEGs)
2.6. Functional Analysis of DEGs
2.7. Validation of Transcriptomics Data
3. Results
3.1. The Four Developmental Stages of Primordium Formation of P. tuoliensis
3.2. RNA Sequencing
3.3. Gene Functional Annotation
3.4. Identification of DEGs in Four Stages
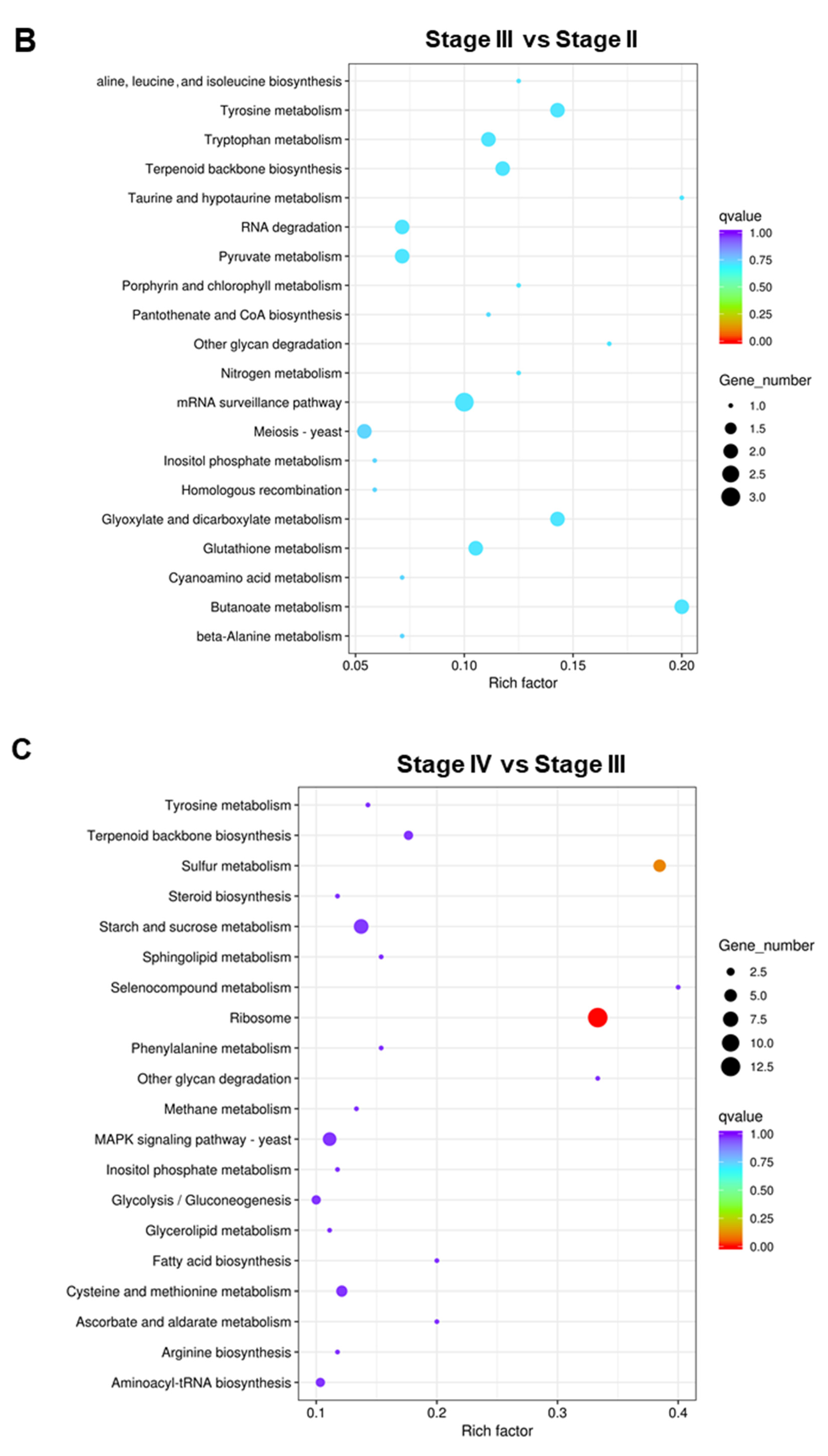
3.5. Functional Enrichment of DEGs
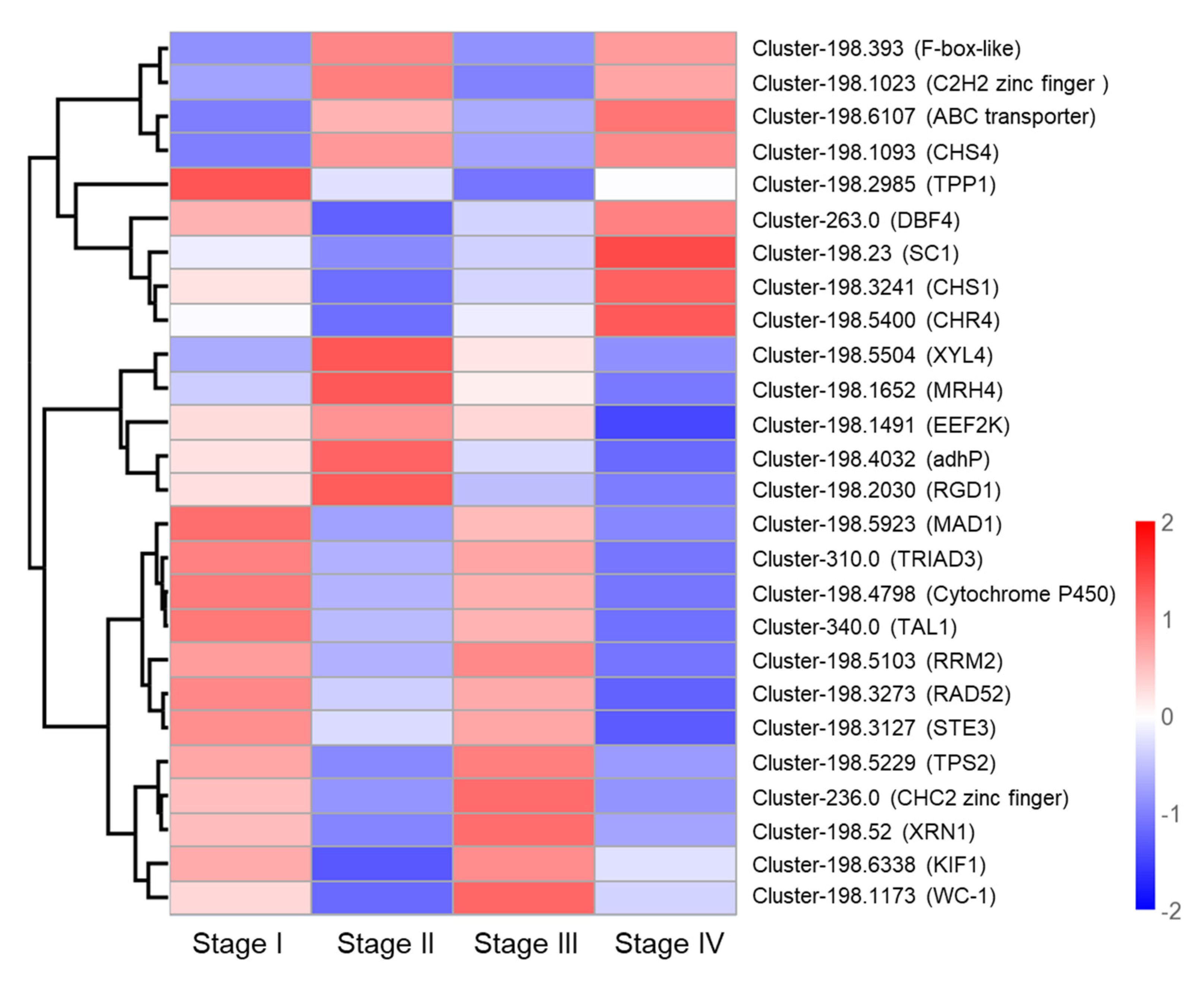
3.6. Identification of Candidate Genes Involved in Primordium Formation
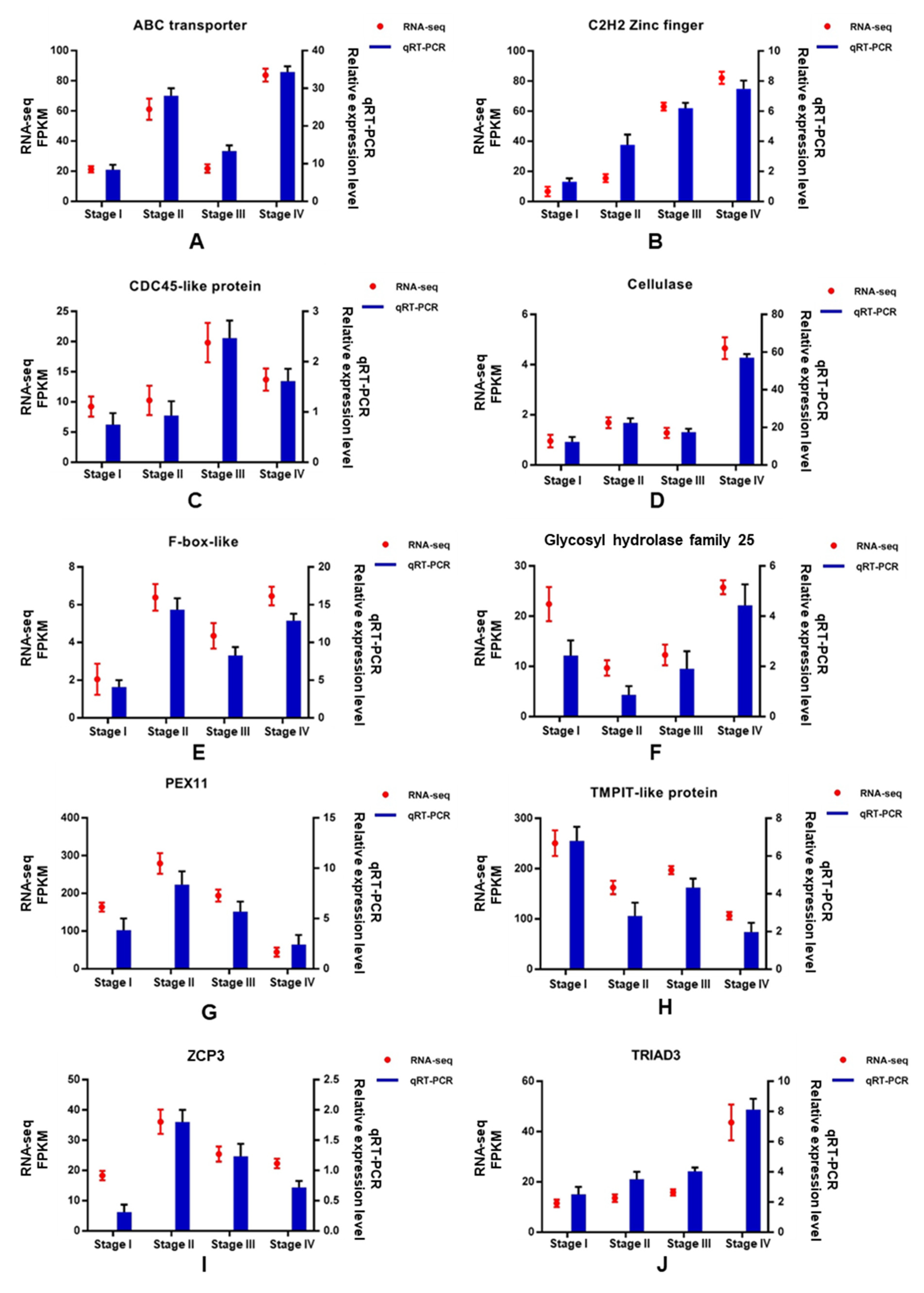
3.7. Validation of DEGs by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, M.; Zhang, J.; Chen, Q.; Wu, X.; Gao, W.; Deng, W.; Huang, C. The famous cultivated mushroom Bailinggu is a separate species of the Pleurotus eryngii species complex. Sci. Rep. 2016, 6, 33066. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zou, Y.; Yu, Y.; Chen, B.; Zheng, Q.; Ye, Z.; Wei, T.; Ye, S.; Guo, L.; Lin, J. Comparative transcriptome and proteome provide new insights into the regulatory mechanisms of the postharvest deterioration of Pleurotus tuoliensis fruitbodies during storage. Food Res. Int. 2021, 147, 110540. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zheng, Q.; Lu, J.; Zou, Y.; Lin, J.; Guo, L.; Ye, S.; Xing, Z. Chemical composition and deterioration mechanism of Pleurotus tuoliensis during postharvest storage. Food Chem. 2021, 338, 127731. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Yoon, K.N.; Lee, J.S.; Cho, H.J.; Lee, T.S. Consequence of the antioxidant activities and tyrosinase inhibitory effects of various extracts from the fruiting bodies of Pleurotus ferulae. Saudi J. Biol. Sci. 2012, 19, 111–118. [Google Scholar] [CrossRef]
- Wang, W.; Chen, K.; Liu, Q.; Johnston, N.; Ma, Z.; Zhang, F.; Zheng, F. Suppression of tumor growth by Pleurotus ferulae ethanol extract through induction of cell apoptosis, and inhibition of cell proliferation and migration. PLoS ONE 2014, 9, e102673. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Aipire, A.; Luo, J.; Yuan, P.; Zhang, F. The combination of Pleurotus ferulae water extract and CpG-ODN enhances the immune responses and antitumor efficacy of HPV peptides pulsed dendritic cell-based vaccine. Vaccine 2016, 34, 3568–3575. [Google Scholar] [CrossRef]
- Wang, C.; Cui, H.; Wang, Y.; Wang, Z.; Li, Z.; Chen, M.; Li, F. Bidirectional immunomodulatory activities of polysaccharides purified from Pleurotus nebrodensis. Inflammation 2014, 37, 83–93. [Google Scholar] [CrossRef]
- Kawai, G.; Babasaki, K.; Neda, H. Taxonomic position of a Chinese Pleurotus “Bai-Ling-Gu”: It belongs to Pleurotus eryngii (DC.:Fr.) Quél. and evolved in dependently in China. Mycoscience 2008, 49, 75–87. [Google Scholar] [CrossRef]
- Alam, N.; Shim, M.; Lee, M.W.; Shin, P.; Yoo, Y.B.; Lee, T.S. Phylogenetic relationship in different commercial strains of Pleurotus nebrodensis based on ITS sequence and RAPD. Mycobiology 2009, 37, 183–188. [Google Scholar] [CrossRef]
- Zou, Y.; Du, F.; Hu, Q.; Yuan, X.; Dai, D.; Zhu, M. Integration of Pleurotus tuoliensis cultivation and biogas production for utilization of lignocellulosic biomass as well as its benefit evaluation. Bioresour. Technol. 2020, 317, 124042. [Google Scholar] [CrossRef]
- Du, F.; Zou, Y.; Hu, Q.; Jing, Y.; Yang, X. Metabolic profiling of Pleurotus tuoliensis during mycelium physiological maturation and exploration on a potential indicator of mycelial maturation. Front. Microbiol. 2019, 9, 3274. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, S.; Huang, Z.; Yin, L.; Hu, J.; Li, J.; Liu, Y.; Rong, C. Development of a highly productive strain of Pleurotus tuoliensis for commercial cultivation by crossbreeding. Sci. Hortic. 2018, 234, 110–115. [Google Scholar] [CrossRef]
- Gao, W.; Qu, J.; Zhang, J.; Sonnenberg, A.; Chen, Q.; Zhang, Y.; Huang, C. A genetic linkage map of Pleurotus tuoliensis integrated with physical mapping of the de novo sequenced genome and the mating type loci. BMC Genom. 2018, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Fan, W.; Xu, Y.; She, D.; Sun, J. Progress of study on Pleurotus nebrodensis. Seed 2007, 26, 55–58. [Google Scholar]
- Wu, X.; Hou, Z.; Huang, C.; Chen, Q.; Gao, W.; Zhang, J. Cloning, purification and characterization of trehalose-6-phosphate synthase from Pleurotus tuoliensis. PeerJ 2018, 6, e5230. [Google Scholar] [CrossRef]
- Wang, G.; Li, M.; Cheng, H.; Zhang, C.; Deng, W.; Li, T. Expression profiling of Cordyceps DnaJ protein family in Tolypocladium guangdongense during developmental and temperature stress processes. Gene 2020, 743, 144563. [Google Scholar] [CrossRef]
- Fu, Y.; Liang, Y.; Dai, Y.; Yang, C.; Duan, M.; Zhang, Z.; Hu, S.; Zhang, Z.; Li, Y. De novo sequencing and transcriptome analysis of Pleurotus eryngii subsp. tuoliensis (Bailinggu) mycelia in response to cold stimulation. Molecules 2016, 21, 560. [Google Scholar] [CrossRef]
- Purschwitz, J.; Müller, S.; Kastner, C.; Fischer, R. Seeing the rain bow: Light sensing in fungi. Curr. Opin. Microbiol. 2006, 9, 566571. [Google Scholar] [CrossRef]
- Jang, M.J.; Lee, Y.H.; Ju, Y.C.; Kim, S.M.; Koo, H.M. Effect of color of light emitting diode on development of fruit body in Hypsizygus marmoreus. Mycobiology 2013, 41, 63–66. [Google Scholar] [CrossRef]
- Xie, C.; Gong, W.; Zhu, Z.; Yan, L.; Hu, Z.; Peng, Y. Comparative transcriptomics of Pleurotus eryngii reveals blue-light regulation of carbohydrate-active enzymes (CAZymes) expression at primordium differentiated into fruiting body stage. Genomics 2018, 110, 201–209. [Google Scholar] [CrossRef]
- Orban, A.; Weber, A.; Herzog, R.; Hennicke, F.; Rühl, M. Transcriptome of different fruiting stages in the cultivated mushroom Cyclocybe aegerita suggests a complex regulation of fruiting and reveals enzymes putatively involved in fungal oxylipin biosynthesis. BMC Genom. 2021, 22, 324. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, Y.; Keerio, A.A.; Ma, A. Identification of hydrophobin genes and their physiological functions related to growth and development in Pleurotus ostreatus. Microbiol. Res. 2021, 247, 126723. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Chen, H.; Zhang, M.; Wen, Q.; Qiu, L.; Shen, J. Identification and expression analysis of Pofst3 suggests a role during Pleurotus ostreatus primordia formation. Fungal Biol. 2019, 3, 123. [Google Scholar] [CrossRef]
- Zhu, W.; Hu, J.; Li, Y.; Yang, B.; Guan, Y.; Xu, C.; Chen, F.; Chi, J.; Bao, Y. Comparative proteomic analysis of Pleurotus ostreatus reveals great metabolic differences in the cap and stipe development and the potential role of Ca2+ in the primordium differentiation. Int. J. Mol. Sci. 2019, 20, 6317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, B.; Wei, W.; Xiong, Y.; Zhu, W.; Peng, F.; Yu, Y.; Zheng, Y.; Chen, P. De novo analysis of Wolfiporia cocos transcriptome to reveal the differentially expressed carbohydrate-active enzymes (CAZymes) genes during the early stage of sclerotial growth. Front. Microbiol. 2016, 7, 83. [Google Scholar] [CrossRef]
- Yin, J.; Xin, X.; Weng, Y.; Gui, Z. Transcriptome-wide analysis reveals the progress of Cordyceps militaris subculture degeneration. PLoS ONE 2017, 12, e0186279. [Google Scholar]
- Ye, D.; Du, F.; Zou, Y.; Hu, Q. Transcriptomics analysis of primordium formation in Pleurotus eryngii. Genes 2021, 12, 1863. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef]
- Du, F.; Ti, N.E.Z.Y.Y.L.M.M.; Hu, Q.; Zou, Y.; Ye, D.; Zhang, A. A comparative transcriptome analysis reveals physiological maturation properties of mycelia in Pleurotus tuoliensis. Genes 2019, 10, 703. [Google Scholar] [CrossRef]
- Liu, X.; Xia, E.; Li, M.; Cui, Y.; Wang, P.; Zhang, J.; Xie, B.; Xu, J.; Yan, J.; Li, J.; et al. Transcriptome data reveal conserved patterns of fruiting body development and response to heat stress in the mushroom-forming fungus Flammulina filiformis. PLoS ONE 2020, 15, e0239890. [Google Scholar] [CrossRef]
- Qiu, Z.; Wu, X.; Zhang, J.; Huang, C. High-temperature induced changes of extracellular metabolites in Pleurotus ostreatus and their positive effects on the growth of Trichoderma asperellum. Front. Microbiol. 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dai, Y.; Yang, C.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.; Li, Y. Comparative transcriptome analysis identified candidate genes related to bailinggu mushroom formation and genetic markers for genetic analyses and breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without are ference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 93–99. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2–DDCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hong, M. Biochemical studies on the structure-function relationship of major drug transporters in the ATP-binding cassette family and solute carrier family. Adv. Drug Deliv. Rev. 2017, 116, 3–20. [Google Scholar] [CrossRef]
- Gou, Y.; He, Y.; Mao, Z. Effect of after-riping time on the fruiting and yield of Pleurotus tuoliensis. Mushroom 2003, 25, 33–34. [Google Scholar]
- Ohm, R.A.; de Jong, J.F.; de Bekker, C.; Wösten, H.A.; Lugones, L.G. Transcription factor genes of Schizophyllum commune involved in regulation of mushroom formation. Mol. Microbiol. 2011, 81, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, J.F.; Patil, M.B.; Gehrmann, T.; Reinders, M.J.; Wösten, H.A.; Lugones, L.G. Transcription factors of Schizophyllum commune involved in mushroom formation and modulation of vegetative growth. Sci. Rep. 2017, 7, 310. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, J.F.; Vos, A.M.; Scholtmeijer, K.; Hendrix, E.; Baars, J.J.; Gehrmann, T.; Reinders, M.J.; Lugones, L.G.; Wösten, H.A. The transcriptional regulator c2h2 accelerates mushroom formation in Agaricus bisporus. Appl. Microbiol. Biotechnol. 2016, 100, 7151–7159. [Google Scholar] [CrossRef] [PubMed]
- Ceccaroli, P.; Buffalini, M.; Saltarelli, R.; Barbieri, E.; Polidori, E.; Ottonello, S.; Kohler, A.; Tisserant, E.; Martin, F.; Stocchi, V. Genomic profiling of carbohydrate metabolism in the ectomycorrhizal fungus Tuber melanosporum. New Phytol. 2011, 189, 751–764. [Google Scholar] [CrossRef]
- Toshiaki, K.; Minoru, K. The role of gene expression regulation in yeast cell cycle pathway. Genome Inform. 2000, 11, 268–269. [Google Scholar]
- Ye, D.; Du, F.; Hu, Q.; Zou, Y.; Bai, X. Transcriptome analysis reveals candidate genes involved in light-induced primordium differentiation in Pleurotus eryngii. Int. J. Mol. Sci. 2021, 23, 435. [Google Scholar] [CrossRef]
- Abdullah, U.; Cullen, P.J. The tRNA modification complex elongator regulates the Cdc42-dependent mitogen-activated protein kinase pathway that controls filamentous growth in yeast. Eukaryot. Cell 2009, 8, 1362–1372. [Google Scholar] [CrossRef]
- Martínez-Soto, D.; Ruiz-Herrera, J. Functional analysis of the MAPK pathways in fungi. Rev. Iberoam. Micol. 2017, 34, 192–202. [Google Scholar] [CrossRef]
- Cheng, C.; Au, C.H.; Wilke, S.K.; Stajich, J.E.; Zolan, M.E.; Pukkila, P.J.; Kwan, H.S. 5′-Serial analysis of gene expression studies reveal a transcriptomic switch during fruiting body development in Coprinopsis cinerea. BMC Genom. 2013, 14, 195. [Google Scholar] [CrossRef]
- Tisch, D.; Schmoll, M. Light regulation of metabolic pathways in fungi. Appl. Microbiol. Biotechnol. 2010, 85, 1259–1277. [Google Scholar] [CrossRef]
- Schwerdtfeger, C.; Linden, H. VIVID is a flavoprotein and serves as a fungal blue light photoreceptor for photoadaptation. EMBO J. 2003, 22, 4846–4855. [Google Scholar] [CrossRef] [PubMed]
- Idnurm, A.; Heitman, J. Light controls growth and development via a conserved pathway in the fungal kingdom. PLoS Biol. 2005, 3, e95. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.J.; Lee, Y.H.; Kim, J.H.; Ju, Y.C. Effect of LED light on primordium formation, morphological properties, ergosterol content and antioxidant activity of fruit body in Pleurotus eryngii. Korean J. Mycol. 2011, 39, 175–179. [Google Scholar] [CrossRef]
- Ye, D.; Du, F.; Zou, Y.; Zhang, H.; Hou, Z.; Hu, Q. Effects of light conditions on the differentiation and physiological effects of Pleurotus eryngii primordium. China J. Appl. Environ. Biol. 2019, 25, 1107–1112. [Google Scholar]
- Wang, H.; Tong, X.; Tian, F.; Jia, C.; Li, C.; Li, Y. Transcriptomic profiling sheds light on the blue-light and red-light response of oyster mushroom (Pleurotus ostreatus). AMB Express 2018, 10, 10. [Google Scholar] [CrossRef]
- Song, T.; Shen, Y.; Jin, Q.; Feng, W.; Fan, L.; Cai, W. Comparative phosphoproteome analysis to identify candidate phosphoproteins involved in blue light-induced brown film formation in Lentinula edodes. PeerJ 2020, 8, e9859. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, R.; Qiu, Y.; Wu, H.; Xiang, Q.; Yu, X.; Zhao, K.; Zhang, X.; Chen, Q.; Penttinen, P.; et al. RNA-seq profiling showed divergent carbohydrate-active enzymes (CAZymes) expression patterns in Lentinula edodes at brown film formation stage under blue light induction. Front. Microbiol. 2020, 11, 1044. [Google Scholar] [CrossRef]
- Yoo, S.I.; Lee, H.Y.; Markkandan, K.; Moon, S.; Ahn, Y.J.; Ji, S.; Ko, J.; Kim, S.J.; Ryu, H.; Hong, C.P. Comparative transcriptome analysis identified candidate genes involved in mycelium browning in Lentinula edodes. BMC Genom. 2019, 20, 121. [Google Scholar] [CrossRef]
- Du, F.; Zou, Y.; Hu, Q.; Zhang, R.; Ye, D. Comparative transcriptomic analysis reveals molecular processes involved in pileus morphogenesis in Pleurotus eryngii under different light conditions. Genomics 2020, 112, 1707–1715. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Zhou, J.; Cao, Z.; Hu, B.; Wang, J.; Guo, J.; Zheng, S. De Novo Assembly Transcriptome Analysis Reveals the Preliminary Molecular Mechanism of Primordium Formation in Pleurotus tuoliensis. Genes 2022, 13, 1747. https://doi.org/10.3390/genes13101747
Wang C, Zhou J, Cao Z, Hu B, Wang J, Guo J, Zheng S. De Novo Assembly Transcriptome Analysis Reveals the Preliminary Molecular Mechanism of Primordium Formation in Pleurotus tuoliensis. Genes. 2022; 13(10):1747. https://doi.org/10.3390/genes13101747
Chicago/Turabian StyleWang, Chunxia, Jinkan Zhou, Zijian Cao, Bao Hu, Jing Wang, Jinying Guo, and Suyue Zheng. 2022. "De Novo Assembly Transcriptome Analysis Reveals the Preliminary Molecular Mechanism of Primordium Formation in Pleurotus tuoliensis" Genes 13, no. 10: 1747. https://doi.org/10.3390/genes13101747
APA StyleWang, C., Zhou, J., Cao, Z., Hu, B., Wang, J., Guo, J., & Zheng, S. (2022). De Novo Assembly Transcriptome Analysis Reveals the Preliminary Molecular Mechanism of Primordium Formation in Pleurotus tuoliensis. Genes, 13(10), 1747. https://doi.org/10.3390/genes13101747