
Comparative Transcriptome and sRNAome Analyses Reveal the Regulatory Mechanisms of Fruit Ripening in a Spontaneous Early-Ripening Navel Orange Mutant and Its Wild Type

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Treatments
2.2. Quantification of Carotenoid, Cellulose, Pectin Substances and Related Enzyme Activities and Endogenous Hormones
2.3. Library Preparation for Transcriptome Sequencing
2.4. Library Preparation for Small RNA Sequencing
2.5. Data Analysis
2.6. RNA Extraction and qRT-PCR Analysis
2.7. Statistical Analyses
3. Results
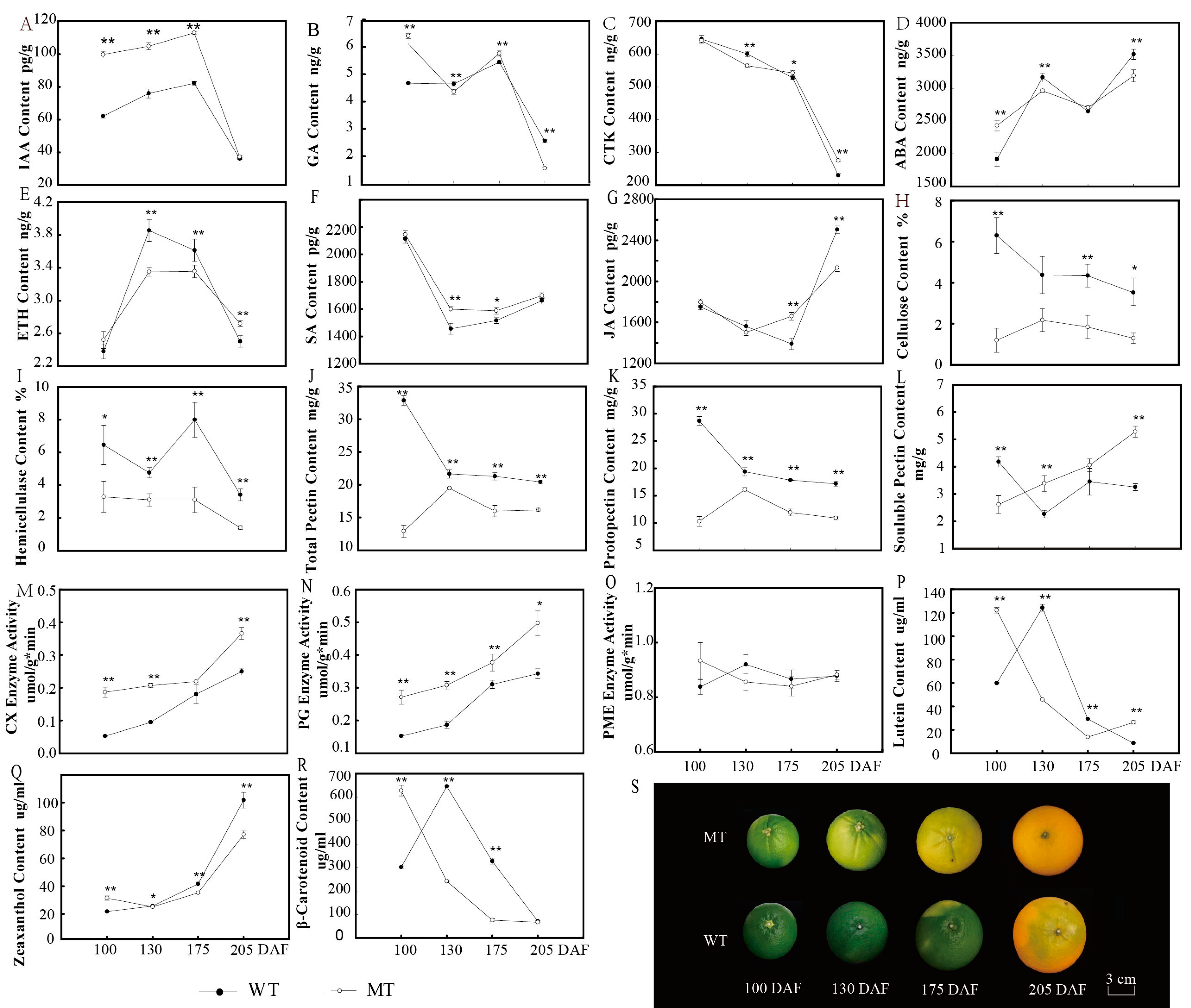
3.1. Changes in Metabolic Materials during Fruit Ripening in MT and WT
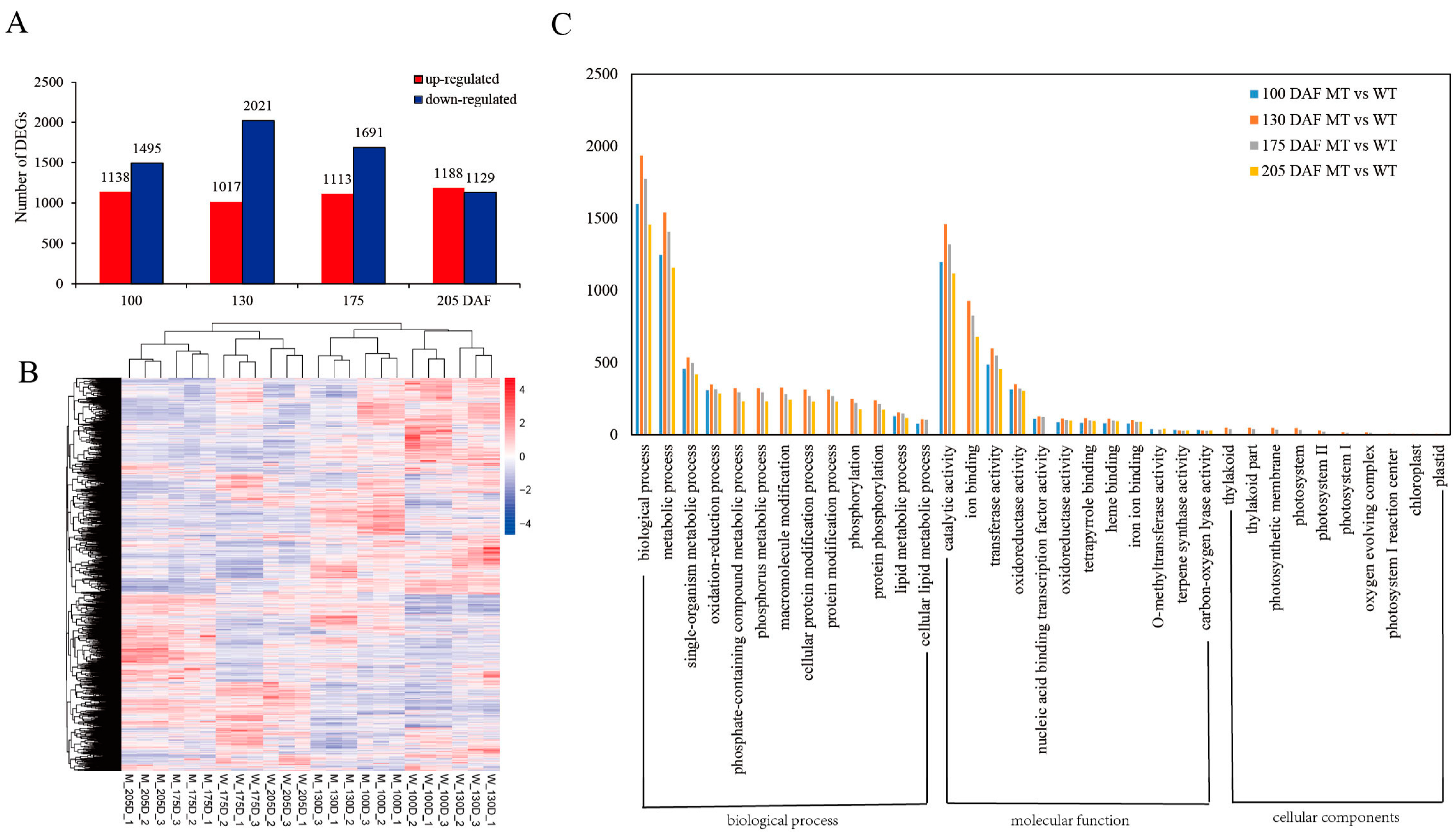
3.2. Fruit Transcriptome Differences between MT and WT during Fruit Ripening
3.2.1. Expression Analysis of Different Genes in MT and WT
3.2.2. Annotation of DEGs in MT and WT
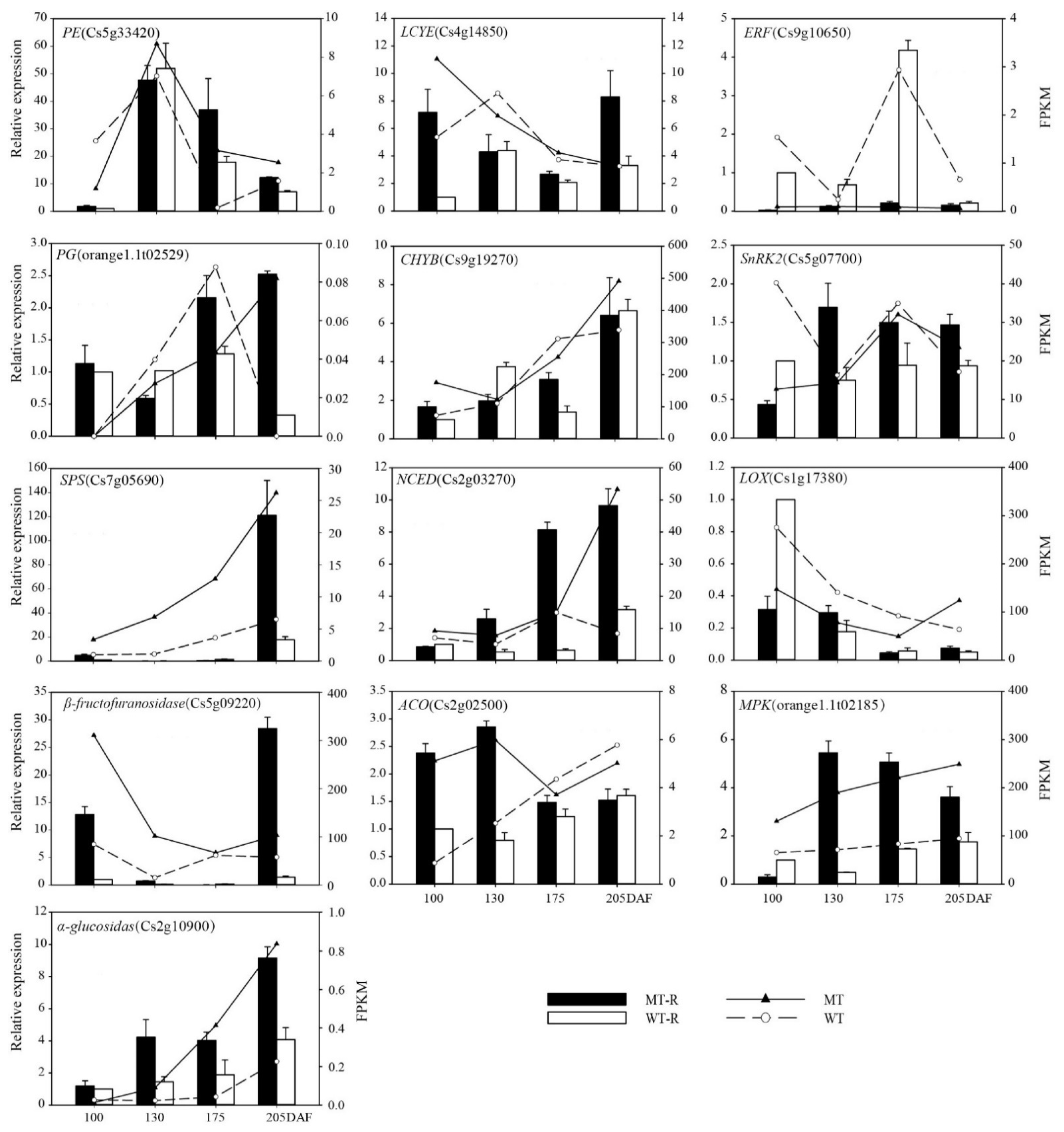
3.2.3. Verification of Differentially Expressed Genes
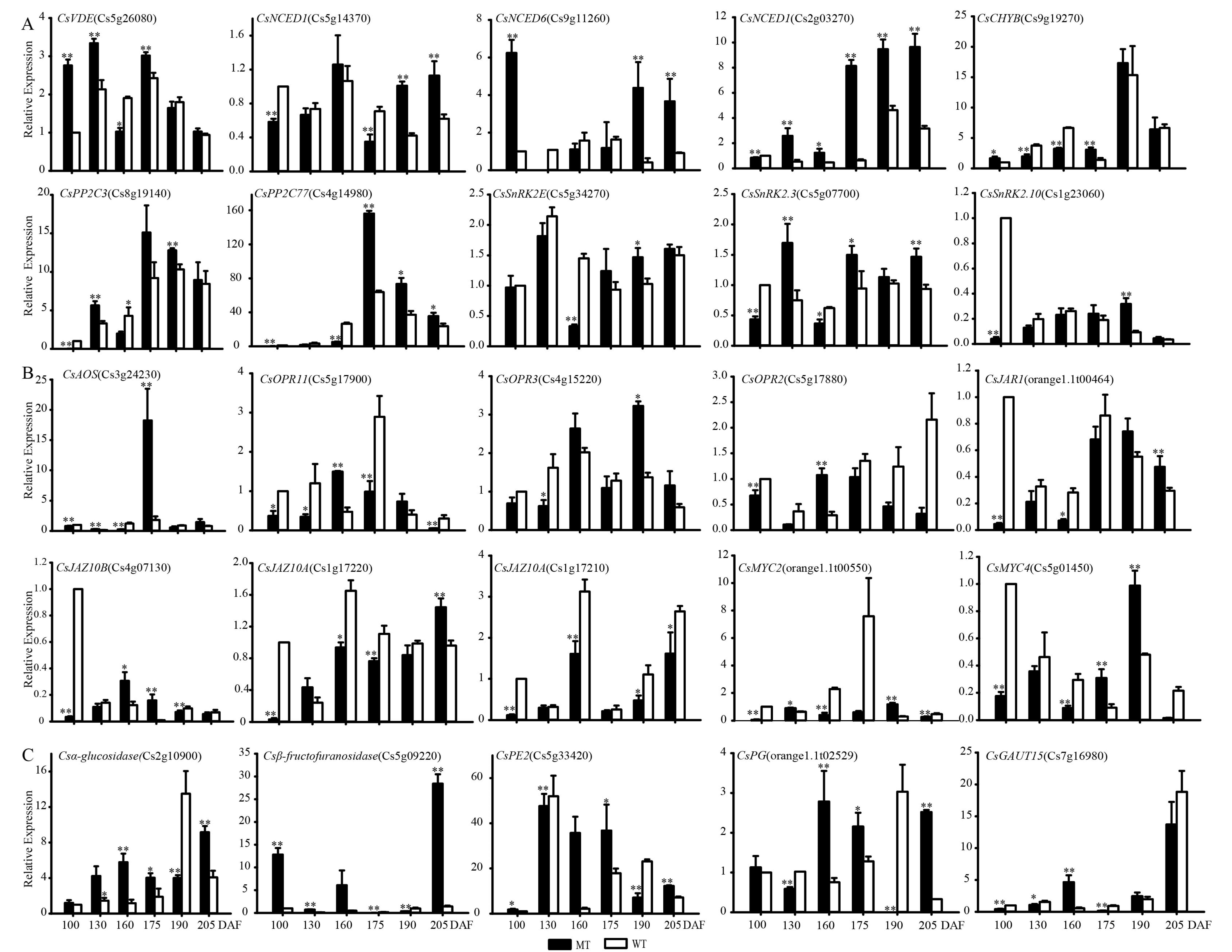
3.2.4. Expression Pattern Comparisons for ABA, JA Synthesis, Signal Transduction and Some Starch and Sucrose Metabolism-Related Genes during Fruit Development and Ripening in MT and WT
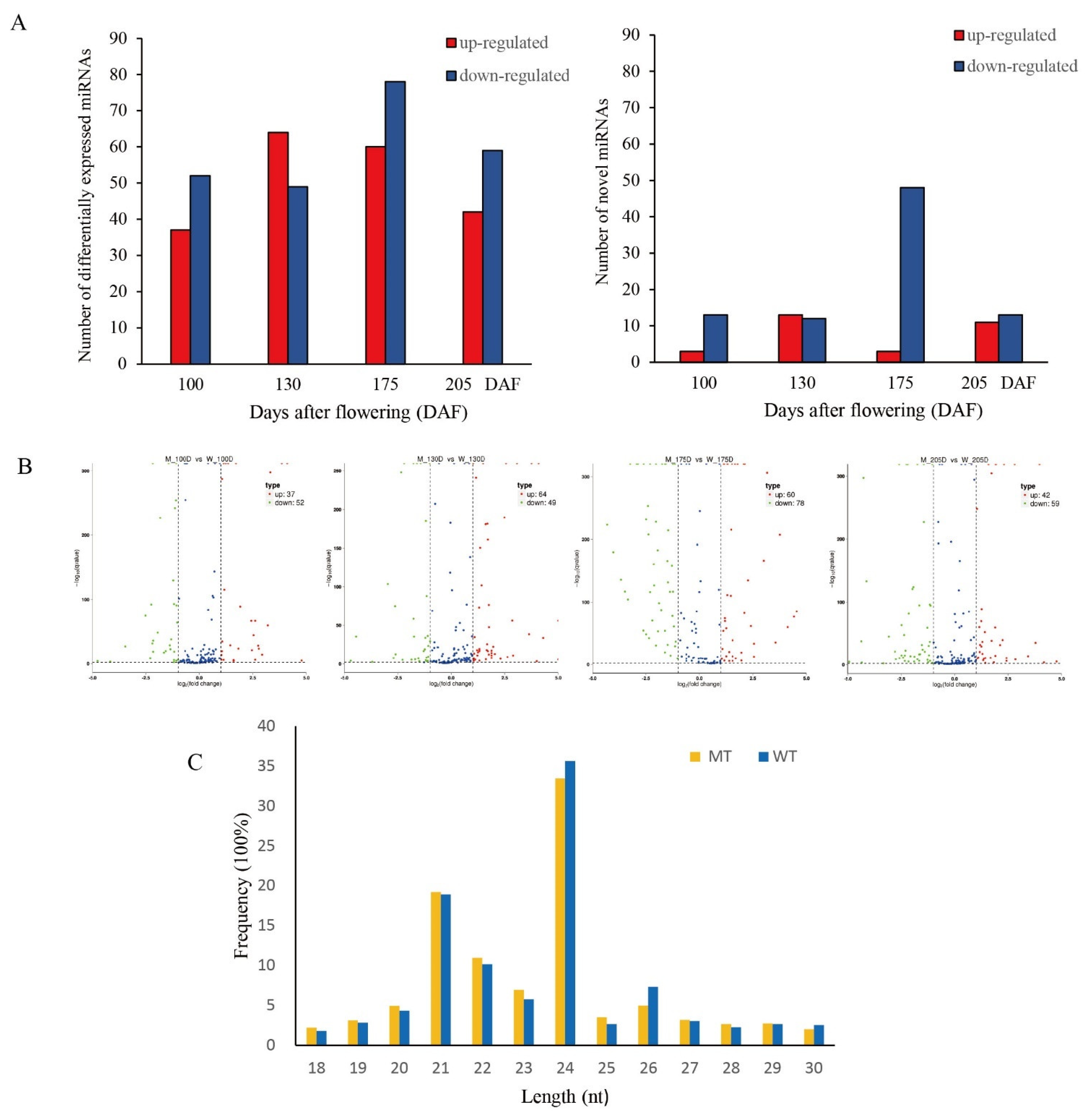
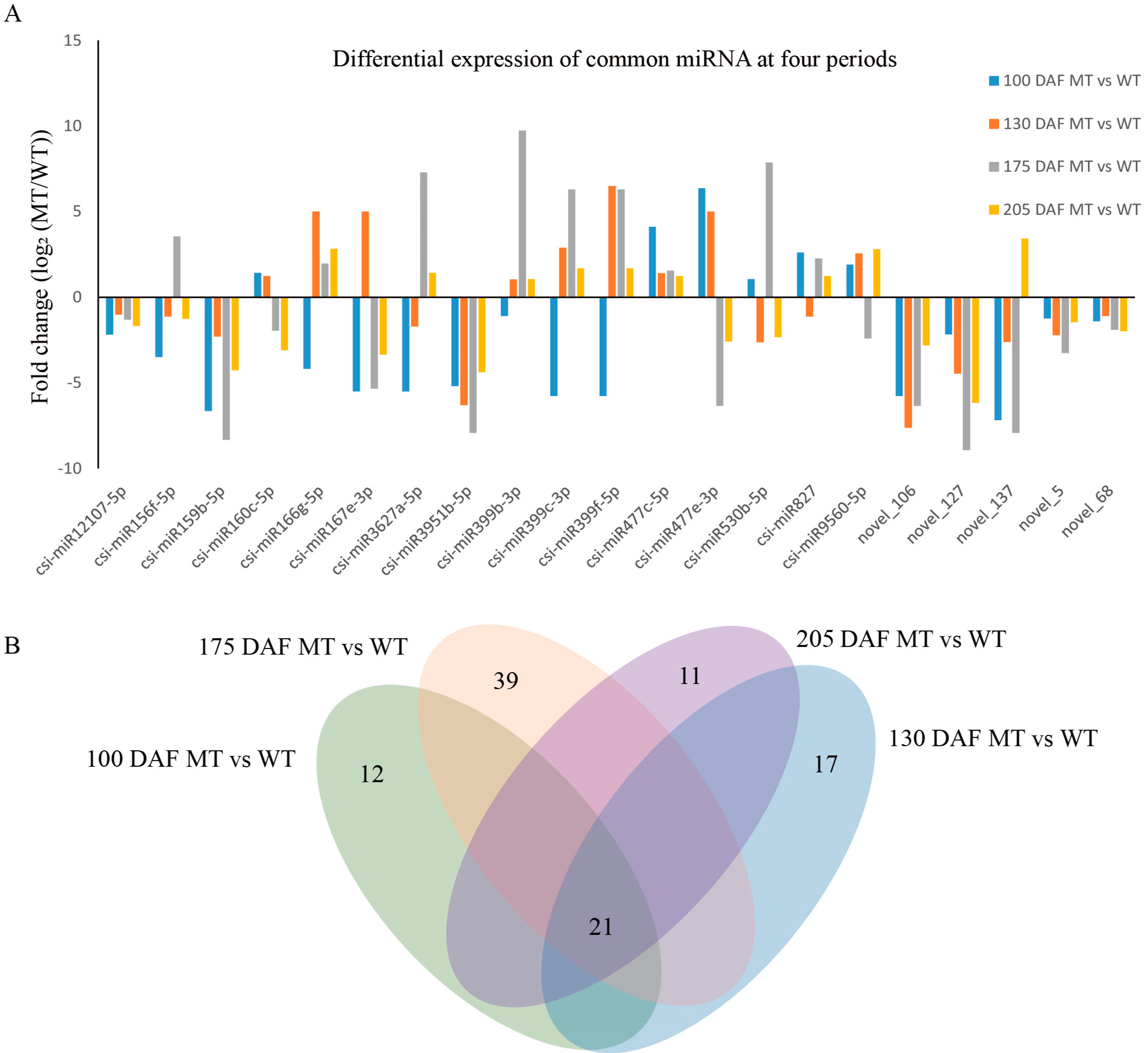
3.3. Differential Expression Analysis of miRNA in MT and WT
3.3.1. Expression Analysis of Known and Novel miRNAs in MT and WT
3.3.2. Prediction and Annotation of Target Genes
3.3.3. Functional Analysis of Target Genes in Which Major miRNAs Were Involved in Fruit Development
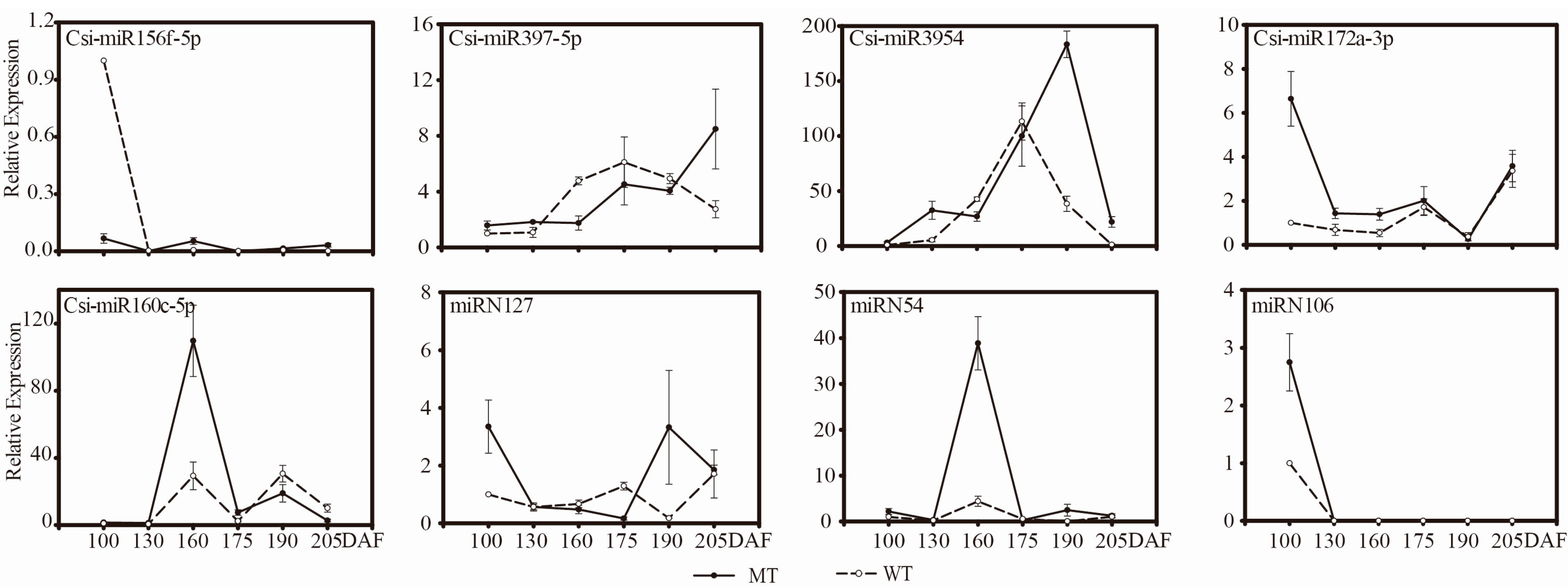
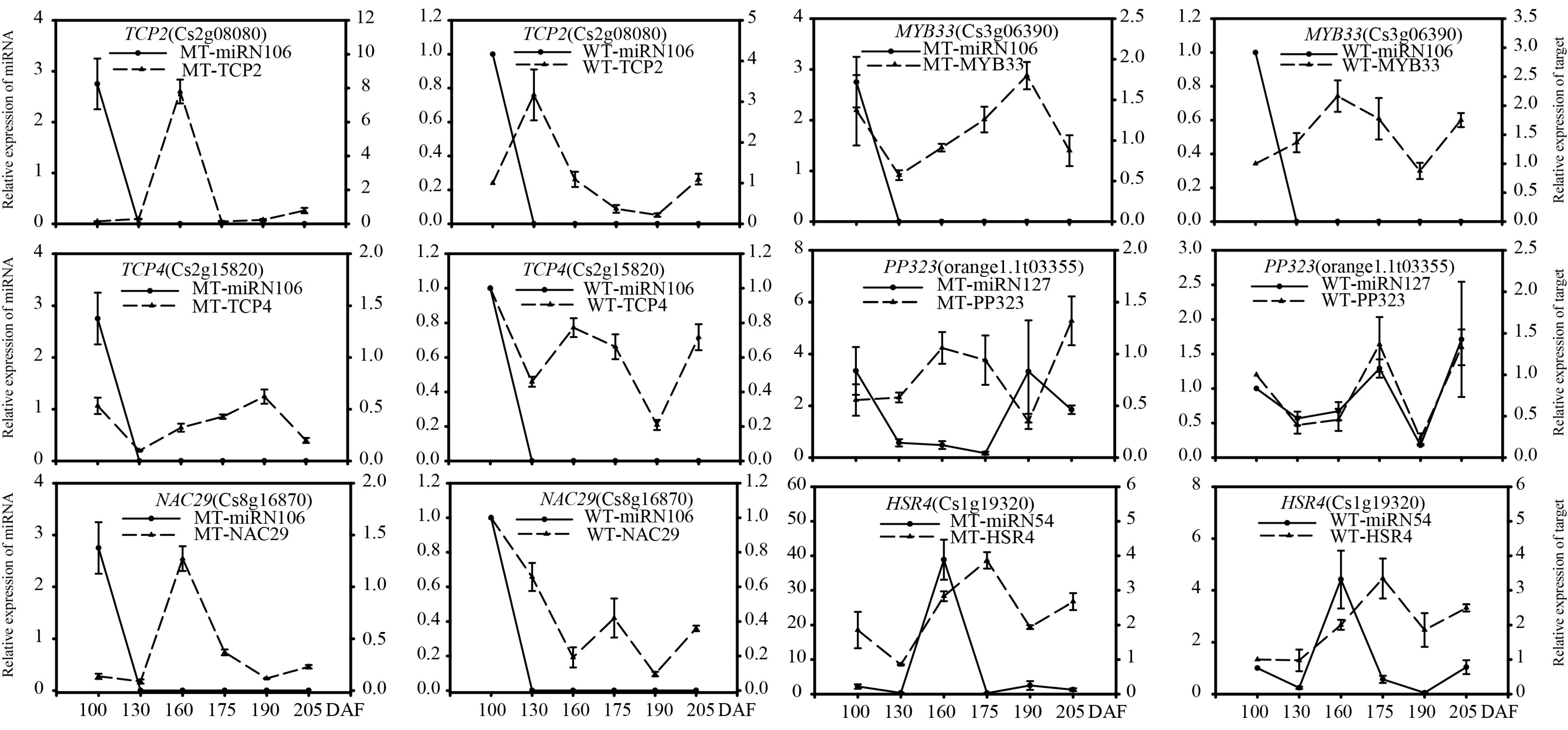
3.3.4. Comparative Expression Patterns of miRNAs and Target Genes in MT and WT
4. Discussion
4.1. Identification of Main Regulators and Metabolic Pathways Involved in Fruit Ripening
4.2. The Early-Ripening Trait Was Regulated by miRNAs in Citrus Fruit
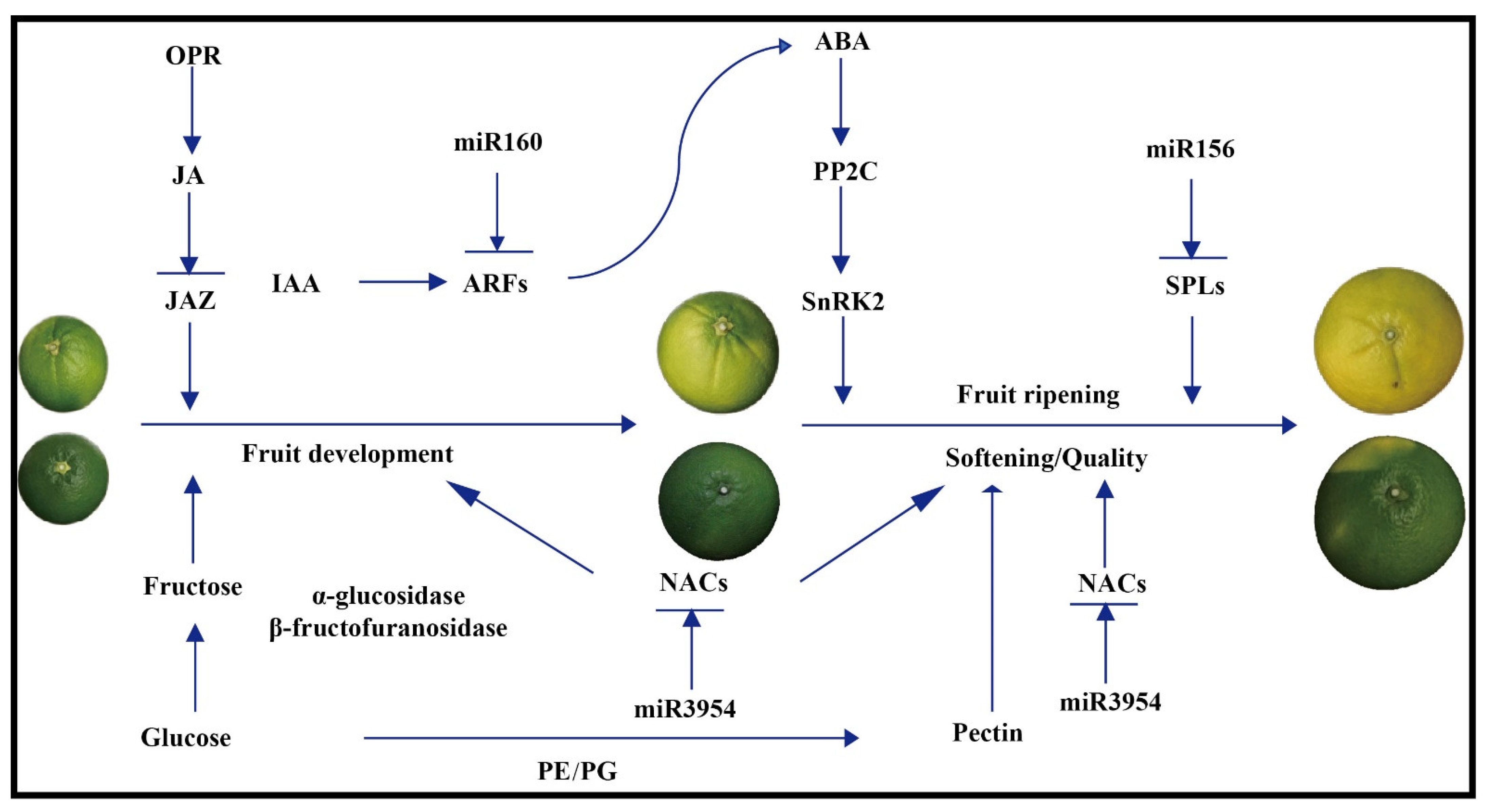
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Iglesias, D.J.; Cercós, M.; Colmenero-Flores, J.M.; Naranjo, M.A.; Ríos, G.; Carrera, E.; Ruiz-Rivero, O.; Lliso, I.; Morillon, R.; Tadeo, F.R.; et al. Physiology of citrus fruiting. Braz. J. Plant Physiol. 2007, 19, 333–362. [Google Scholar] [CrossRef]
- Bouzayen, M.; Latché, A.; Nath, P.; Pech, J.C. Mechanism of Fruit Ripening. In Plant Developmental Biology-Biotechnological Perspectives; Springer: Berlin/Heidelberg, Germany, 2010; Volume 1, pp. 319–339. [Google Scholar]
- Liu, Y.; Liu, Q.; Xiong, J.; Deng, X. Difference of a citrus late-ripening mutant (Citrus sinensis) from its parental line in sugar and acid metabolism at the fruit ripening stage. Sci. China Ser. C Life Sci. 2007, 50, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, Z.; Zhang, Y.; Chai, L.; Yi, H.; Deng, X. An integrative analysis of the transcriptome and proteome of the pulp of a spontaneous late-ripening sweet orange mutant and its wild type improves our understanding of fruit ripening in citrus. J. Exp. Bot. 2014, 65, 1651–1671. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Fu, L.; Yi, H. Genome-Wide Identification of the Transcription Factors Involved in Citrus Fruit Ripening from the Transcriptomes of a Late-Ripening Sweet Orange Mutant and Its Wild Type. PLoS ONE 2016, 11, e0154330. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zheng, S.; Feng, G.; Yi, H. Comparative Analysis of miRNAs and Their Target Transcripts between a Spontaneous Late-Ripening Sweet Orange Mutant and Its Wild-Type Using Small RNA and Degradome Sequencing. Front. Plant Sci. 2016, 7, 1416. [Google Scholar] [CrossRef]
- Alos, E.; Distefano, G.; Rodrigo, M.J.; Gentile, A.; Zacarias, L. Altered sensitivity to ethylene in ‘Tardivo’, a late-ripening mutant of Clementine mandarin. Physiol. Plant. 2014, 151, 507–521. [Google Scholar] [CrossRef]
- Terol, J.; Nueda, M.J.; Ventimilla, D.; Tadeo, F.; Talon, M. Transcriptomic analysis of Citrus clementina mandarin fruits maturation reveals a MADS-box transcription factor that might be involved in the regulation of earliness. BMC Plant Biol. 2019, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Wang, X.J.; Wu, J.X.; Chen, S.Y.; Chen, H.; Chai, L.J.; Yi, H.L. Comparative transcriptome analyses between a spontaneous late-ripening sweet orange mutant and its wild type suggest the functions of ABA, sucrose and JA during citrus fruit ripening. PLoS ONE 2014, 9, e116056. [Google Scholar] [CrossRef]
- Rodrigo, M.J.; Marcos, J.F.; Alferez, F.; Mallent, M.D.; Zacarias, L. Characterization of Pinalate, a novel Citrus sinensis mutation with a fruit-specific alteration that results in yellow pigmentation and decreased ABA content. J. Exp. Bot. 2003, 54, 727–738. [Google Scholar] [CrossRef]
- Rodrigo, M.J.; Alquezar, B.; Zacarias, L. Cloning and characterization of two 9-cis-epoxycarotenoid dioxygenase genes, differentially regulated during fruit maturation and under stress conditions, from orange (Citrus sinensis L. Osbeck). J. Exp. Bot. 2006, 57, 633–643. [Google Scholar] [CrossRef]
- Romero, P.; Lafuenta, M.T.; Rodrigo, M.J. The Citrus ABA signalosome: Identification and transcriptional regulation during sweet orange fruit ripening and leaf dehydration. J. Exp. Bot. 2012, 63, 4931–4945. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Yu, H.; Xu, Q.; Deng, X. Transcriptomic analysis of differentially expressed genes in an orange-pericarp mutant and wild type in pummelo (Citrus grandis). BMC Plant Biol. 2015, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, Y.; Zhu, A.; Wu, X.; Ye, J.; Yu, K.; Guo, W.; Deng, X. Discovery and comparative profiling of microRNAs in a sweet orange red-flesh mutant and its wild type. BMC Genom. 2010, 11, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Yu, K.; Zhu, A.; Ye, J.; Liu, Q.; Zhang, J.; Deng, X. Comparative transcripts profiling reveals new insight into molecular processes regulating lycopene accumulation in a sweet orange (Citrus sinensis) red-flesh mutant. BMC Genom. 2009, 10, 540. [Google Scholar] [CrossRef]
- Wang, J.H.; Liu, J.J.; Chen, K.L.; Li, H.W.; He, J.; Guan, B.; He, L. Comparative transcriptome and proteome profiling of two Citrus sinensis cultivars during fruit development and ripening. BMC Genom. 2017, 18, 984. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, Y.; Zhu, K.; Yang, W.; Ye, J.; Chai, L.; Xu, Q.; Deng, X. The Citrus Transcription Factor CsMADS6 Modulates Carotenoid Metabolism by Directly Regulating Carotenogenic Genes. Plant Physiol. 2018, 176, 2657–2676. [Google Scholar] [CrossRef]
- Hou, B.Z.; Li, C.L.; Han, Y.Y.; Shen, Y.Y. Characterization of the hot pepper (Capsicum frutescens) fruit ripening regulated by ethylene and ABA. BMC Plant Biol. 2018, 18, 162. [Google Scholar] [CrossRef]
- Song, M.; Wang, S.; Chai, L.; Zhang, S.; Shen, Y. Characterization of an ABA-Induced and K+ Channel Gene FaKAT1 that Regulates Strawberry Fruit Ripening. J. Plant Growth Regul. 2016, 36, 312–322. [Google Scholar] [CrossRef]
- Chen, C.; Zeng, Z.; Liu, Z.; Xia, R. Small RNAs, emerging regulators critical for the development of horticultural traits. Hortic. Res. 2018, 5, 63. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Chen, D.; Wu, X.; Huang, D.; Chen, L.; Li, L.; Deng, X.; Xu, Q. Genome-wide comparison of microRNAs and their targeted transcripts among leaf, flower and fruit of sweet orange. BMC Genom. 2014, 15, 3–15. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, J.; Liu, Y.; Zhao, X.; Deng, X.; Guo, L.; Gu, J. A novel bud mutation that confers abnormal patterns of lycopene accumulation in sweet orange fruit (Citrus sinensis L. Osbeck). J. Exp. Bot. 2007, 58, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jiang, T.; Yao, J. A Two-Step Chemical Process for the Extraction of Cellulose Fiber and Pectin from Mulberry Branch Bark Efficiently. J. Polym. Environ. 2011, 19, 568–573. [Google Scholar] [CrossRef]
- Taylor, K.A. A Colorirnetric Method for the Quantitation of Galacturonic Acid. Appl. Biochem. Biolechnology 1993, 43, 51–54. [Google Scholar] [CrossRef]
- Mongeau, R.; Brassard, R. Determination of Neutral Detergent Fiber in Breakfast Cereals Pentose, Hemicellulose, Cellulose and Lignin Content. J. Food Sci. 1982, 47, 550–555. [Google Scholar] [CrossRef]
- Lohani, S.; Trivedi, P.K.; Nath, P. Changes in activities of cell wall hydrolases during ethylene-induced ripening in banana: Effect of 1-MCP, ABA and IAA. Postharvest Biol. Technol. 2004, 31, 119–126. [Google Scholar] [CrossRef]
- Pathak, N.; Sanwal, G.G. Multiple forms of polygalacturonase from banana fruits. Phytochemistry 1998, 48, 249–255. [Google Scholar] [CrossRef]
- Abu-Goukh, A.-B.; Bashir, H.A. Changes in pectic enzymes and cellulase activity during guava fruit ripening. Food Chem. 2003, 83, 213–218. [Google Scholar] [CrossRef]
- Lu, H.; Tian, H.; Zhang, M.; Liu, Z.; Chen, Q.; Guan, R.; Wang, H. Water Polishing improved controlled-release characteristics and fertilizer efficiency of castor oil-based polyurethane coated diammonium phosphate. Sci. Rep. 2020, 10, 5763. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, W.; Qiu, L.; Pan, T.; Zheng, W.; Kong, B.; Wang, H.; Li, C.; Liu, Z.; Zhang, M. Physiological-biochemical responses of wheat to blending controlled-release potassium chloride and soluble potassium chloride. Soil Tillage Res. 2021, 212, 105058. [Google Scholar] [CrossRef]
- Qu, Z.; Qi, X.; Liu, Y.; Liu, K.; Li, C. Interactive effect of irrigation and polymer-coated potassium chloride on tomato production in a greenhouse. Agric. Water Manag. 2020, 235, 106149. [Google Scholar] [CrossRef]
- Kumar, R.; Khurana, A.; Sharma, A.K. Role of plant hormones and their interplay in development and ripening of fleshy fruits. J. Exp. Bot. 2014, 65, 4561–4575. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Pei, Y.L.; Liang, B.; Zhang, Y.S.; Zhai, X.W.; He, S.H.; Kai, W.B.; Sun, Y.F.; Leng, P. Role of abscisic acid in regulating fruit set and ripening in squash (Cucurbita pepo L.). N. Z. J. Crop Hortic. Sci. 2016, 44, 274–290. [Google Scholar] [CrossRef]
- Jia, H.F.; Chai, Y.M.; Li, C.L.; Lu, D.; Luo, J.J.; Qin, L.; Shen, Y.Y. Abscisic acid plays an important role in the regulation of strawberry fruit ripening. Plant Physiol. 2011, 157, 188–199. [Google Scholar] [CrossRef]
- Sun, L.; Sun, Y.; Zhang, M.; Wang, L.; Ren, J.; Cui, M.; Wang, Y.; Ji, K.; Li, P.; Li, Q.; et al. Suppression of 9-cis-epoxycarotenoid dioxygenase, which encodes a key enzyme in abscisic acid biosynthesis, alters fruit texture in transgenic tomato. Plant Physiol. 2012, 158, 283–298. [Google Scholar] [CrossRef]
- Ghorbel, M.; Brini, F.; Sharma, A.; Landi, M. Role of jasmonic acid in plants: The molecular point of view. Plant Cell Rep. 2021, 40, 1471–1494. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Feng, Y.; Yuan, S.; Zhao, X.; Wu, C.; Wang, C.; Xue, Z. Different regulatory mechanisms of plant hormones in the ripening of climacteric and non-climacteric fruits: A review. Plant Mol. Biol. 2021, 107, 477–497. [Google Scholar] [CrossRef]
- Min, D.; Li, Z.; Ai, W.; Li, J.; Zhou, J.; Zhang, X.; Mu, D.; Li, F.; Li, X.; Guo, Y. The Co-regulation of Ethylene Biosynthesis and Ascorbate-Glutathione Cycle by Methy Jasmonate Contributes to Aroma Formation of Tomato Fruit during Postharvest Ripening. J. Agric. Food Chem. 2020, 68, 10822–10832. [Google Scholar] [CrossRef]
- Guo, D.-L.; Xi, F.-F.; Yu, Y.-H.; Zhang, X.-Y.; Zhang, G.-H.; Zhong, G.-Y. Comparative RNA-Seq profiling of berry development between table grape ‘Kyoho’ and its early-ripening mutant ’Fengzao’. BMC Genom. 2016, 17, 795–812. [Google Scholar] [CrossRef]
- Feng, G.; Wu, J.; Xu, Y.; Lu, L.; Yi, H. High-spatiotemporal-resolution transcriptomes provide insights into fruit development and ripening in Citrus sinensis. Plant Biotechnol. J. 2021, 19, 1337–1353. [Google Scholar] [CrossRef]
- Giovannoni, J.J. Genetic regulation of fruit development and ripening. Plant Cell 2004, 16, S170–S180. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, D.; Zhang, C.; Xia, X.; Yin, W.; Tian, Q. A Putative PP2C-Encoding Gene Negatively Regulates ABA Signaling in Populus euphratica. PLoS ONE 2015, 10, e0139466. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Murata, M.; Minami, H.; Yamamoto, S.; Kagaya, Y.; Hobo, T.; Yamamoto, A.; Hattori, T. Abscisic acid-activated SNRK2 protein kinases function in the gene-regulation pathway of ABA signal transduction by phosphorylating ABA response element-binding factors. Plant J. 2005, 44, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Tajdel, M.; Mituła, F.; Ludwików, A. Regulation of Arabidopsis MAPKKK18 by ABI1 and SnRK2, components of the ABA signaling pathway. Plant Signal. Behav. 2016, 11, e1139277. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, C.; Yan, Z.; Li, J.; Wang, Y. The methyl jasmonate accelerates the strawberry fruits ripening process. Sci. Hortic. 2019, 249, 250–256. [Google Scholar] [CrossRef]
- Ju, Y.-L.; Liu, B.-C.; Xu, X.-L.; Wu, J.-R.; Sun, W.; Fang, Y.-L. Targeted metabolomic and transcript level analysis reveals the effects of exogenous strigolactone and methyl jasmonate on grape quality. Sci. Hortic. 2022, 299, 111009. [Google Scholar] [CrossRef]
- Li, W.; Liu, B.; Yu, L.; Feng, D.; Wang, H.; Wang, J. Phylogenetic analysis, structural evolution and functional divergence of the 12-oxo-phytodienoate acid reductase gene family in plants. BMC Evol. Biol. 2009, 9, 90. [Google Scholar] [CrossRef]
- Li, W.; Zhou, F.; Liu, B.; Feng, D.; He, Y.; Qi, K.; Wang, H.; Wang, J. Comparative characterization, expression pattern and function analysis of the 12-oxo-phytodienoic acid reductase gene family in rice. Plant Cell Rep. 2011, 30, 981–995. [Google Scholar] [CrossRef]
- Schaller, F.; Biesgen, C.; Müssig, C.; Altmann, T.; Weiler, E.W. 12-Oxophytodienoate reductase 3 (OPR3) is the isoenzyme involved in jasmonate biosynthesis. Planta 2000, 210, 979–984. [Google Scholar] [CrossRef]
- Schemberger, M.O.; Stroka, M.A.; Reis, L.; de Souza Los, K.K.; de Araujo, G.A.T.; Sfeir, M.Z.T.; Galvao, C.W.; Etto, R.M.; Baptistao, A.R.G.; Ayub, R.A. Transcriptome profiling of non-climacteric ‘yellow’ melon during ripening: Insights on sugar metabolism. BMC Genom. 2020, 21, 262. [Google Scholar] [CrossRef]
- Pan, H.; Lyu, S.; Chen, Y.; Xu, S.; Ye, J.; Chen, G.; Wu, S.; Li, X.; Chen, J.; Pan, D. MicroRNAs and Transcripts Associated with an Early Ripening Mutant of Pomelo (Citrus grandis Osbeck). Int. J. Mol. Sci. 2021, 22, 9348. [Google Scholar] [CrossRef]
- Razzaq, K.; Singh, Z.; Khan, A.S.; Khan, S.A.K.U.; Ullah, S. Role of 1-MCP in regulating ‘Kensington Pride’ mango fruit softening and ripening. Plant Growth Regul. 2015, 78, 401–411. [Google Scholar] [CrossRef]
- Chen, J.; Duan, Y.; Hu, Y.; Li, W.; Sun, D.; Hu, H.; Xie, J. Transcriptome analysis of atemoya pericarp elucidates the role of polysaccharide metabolism in fruit ripening and cracking after harvest. BMC Plant Biol. 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Brummell, D.A.; Harpster, M.H. Cell wall metabolism in fruit softening and quality and its manipulation in transgenic plants. Plant Mol. Biol. 2001, 47, 311–340. [Google Scholar]
- Chen, X.; Rechavi, O. Plant and animal small RNA communications between cells and organisms. Nat. Rev. Mol. Cell Biol. 2022, 23, 185–203. [Google Scholar] [CrossRef]
- Cardoso, T.C.S.; Alves, T.C.; Caneschi, C.M.; Santana, D.; Fernandes-Brum, C.N.; Reis, G.L.D.; Daude, M.M.; Ribeiro, T.H.C.; Gomez, M.M.D.; Lima, A.A.; et al. New insights into tomato microRNAs. Sci. Rep. 2018, 8, 16069. [Google Scholar] [CrossRef]
- Liu, D.; Mewalal, R.; Hu, R.; Tuskan, G.A.; Yang, X. New technologies accelerate the exploration of non-coding RNAs in horticultural plants. Hortic. Res. 2017, 4, 17031. [Google Scholar] [CrossRef]
- Zuo, J.; Wang, Y.; Zhu, B.; Luo, Y.; Wang, Q.; Gao, L. Network analysis of noncoding RNAs in pepper provides insights into fruit ripening control. Sci. Rep. 2019, 9, 8734. [Google Scholar] [CrossRef]
- Jose Ripoll, J.; Bailey, L.J.; Mai, Q.A.; Wu, S.L.; Hon, C.T.; Chapman, E.J.; Ditta, G.S.; Estelle, M.; Yanofsky, M.F. microRNA regulation of fruit growth. Nat. Plants 2015, 1, 15036. [Google Scholar] [CrossRef]
- Wei, S.; Gruber, M.Y.; Yu, B.; Gao, M.J.; Khachatourians, G.G.; Hegedus, D.D.; Hannoufa, A. Arabidopsis mutant sk156 reveals complex regulation of SPL15 in a miR156-controlled gene network. BMC Plant Biol. 2012, 12, 1471–2229. [Google Scholar]
- Liu, M.-Y.; Wu, X.-M.; Long, J.-M.; Guo, W.-W. Genomic characterization of miR156 and SQUAMOSA promoter binding protein-like genes in sweet orange (Citrus sinensis). Plant Cell Tissue Organ Cult. 2017, 130, 103–116. [Google Scholar] [CrossRef]
- Cui, M.; Wang, C.; Zhang, W.; Pervaiz, T.; Haider, M.S.; Tang, W.; Fang, J. Characterization of Vv-miR156: Vv-SPL pairs involved in the modulation of grape berry development and ripening. Mol. Genet. Genom. MGG 2018, 293, 1333–1354. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Q.; Zhu, X.; Cui, M.; Jia, H.; Zhang, W.; Tang, W.; Leng, X.; Shen, W. Characterization on the conservation and diversification of miRNA156 gene family from lower to higher plant species based on phylogenetic analysis at the whole genomic level. Funct. Integr. Genom. 2019, 19, 933–952. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ye, Y.; Xu, M.; Feng, L.; Xu, L.A. Roles of the SPL gene family and miR156 in the salt stress responses of tamarisk (Tamarix chinensis). BMC Plant Biol. 2019, 19, 370. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, W.; Wang, X.; Yang, R.; Wu, Z.; Wang, H.; Wang, L.; Hu, Z.; Guo, S.; Zhang, H.; et al. MiR156 regulates anthocyanin biosynthesis through SPL targets and other microRNAs in poplar. Hortic. Res. 2020, 7, 118. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, H.; Hou, Z.; Xu, L.; Jin, W.; Liang, Z. Overexpression of Ath-MIR160b increased the biomass while reduced the content of tanshinones in Salvia miltiorrhiza hairy roots by targeting ARFs genes. Plant Cell Tissue Organ Cult 2020, 142, 327–338. [Google Scholar] [CrossRef]
- Diao, D.; Hu, X.; Guan, D.; Wang, W.; Yang, H.; Liu, Y. Genome-wide identification of the ARF (auxin response factor) gene family in peach and their expression analysis. Mol. Biol. Rep. 2020, 47, 4331–4344. [Google Scholar] [CrossRef]
- Damodharan, S.; Zhao, D.; Arazi, T. A common miRNA160-based mechanism regulates ovary patterning, floral organ abscission and lamina outgrowth in tomato. Plant J. Cell Mol. Biol. 2016, 86, 458–471. [Google Scholar] [CrossRef]
- Gangadhar, B.H.; Venkidasamy, B.; Samynathan, R.; Saranya, B.; Chung, I.-M.; Thiruvengadam, M. Overview of miRNA biogenesis and applications in plants. Biologia 2021, 76, 2309–2327. [Google Scholar] [CrossRef]
- Lin, Y.; Lai, Z.; Tian, Q.; Lin, L.; Lai, R.; Yang, M.; Zhang, D.; Chen, Y.; Zhang, Z. Endogenous target mimics down-regulate miR160 mediation of ARF10, -16, and -17 cleavage during somatic embryogenesis in Dimocarpus longan Lour. Front. Plant Sci. 2015, 6, 956. [Google Scholar] [CrossRef]
- Wojcik, A.M.; Nodine, M.D.; Gaj, M.D. miR160 and miR166/165 Contribute to the LEC2-Mediated Auxin Response Involved in the Somatic Embryogenesis Induction in Arabidopsis. Front. Plant Sci. 2017, 8, 2024. [Google Scholar] [CrossRef]
- Sagar, M.; Chervin, C.; Mila, I.; Hao, Y.; Roustan, J.P.; Benichou, M.; Gibon, Y.; Biais, B.; Maury, P.; Latche, A.; et al. SlARF4, an auxin response factor involved in the control of sugar metabolism during tomato fruit development. Plant Physiol. 2013, 161, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, F.; Tang, Y.; Yuan, Y.; Deng, W.; Li, Z. Auxin Response Gene SlARF3 Plays Multiple Roles in Tomato Development and is Involved in the Formation of Epidermal Cells and Trichomes. Plant Cell Physiol. 2015, 56, 2110–2124. [Google Scholar] [PubMed]
- Liu, S.; Zhang, Y.; Feng, Q.; Qin, L.; Pan, C.; Lamin-Samu, A.T.; Lu, G. Tomato AUXIN RESPONSE FACTOR 5 regulates fruit set and development via the mediation of auxin and gibberellin signaling. Sci. Rep. 2018, 8, 2971. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, W.; Sun, Y.; Chang, H.; Ma, K.; Zhao, Z.; Lu, L. Identification and Expression Analysis of miR160 and Their Target Genes in Cucumber. Biochem. Genet. 2022, 60, 127–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, T.; Dong, X.; Liu, Y.; Liu, Z.; Shi, Z.; Wang, Y.; Qi, M.; Li, T. The role of gibberellins and auxin on the tomato cell layers in pericarp via the expression of ARFs regulated by miRNAs in fruit set. Acta Physiol. Plant. 2016, 38, 77. [Google Scholar] [CrossRef]
- Liu, M.; Dong, H.; Wang, M.; Liu, Q. Evolutionary divergence of function and expression of laccase genes in plants. J. Genet. 2020, 99, 23. [Google Scholar] [CrossRef]
- Xu, X.; Zhou, Y.; Wang, B.; Ding, L.; Wang, Y.; Luo, L.; Zhang, Y.; Kong, W. Genome-wide identification and characterization of laccase gene family in Citrus sinensis. Gene 2019, 689, 114–123. [Google Scholar] [CrossRef]
- Swetha, C.; Basu, D.; Pachamuthu, K.; Tirumalai, V.; Nair, A.; Prasad, M.; Shivaprasad, P.V. Major Domestication-Related Phenotypes in Indica Rice Are Due to Loss of miRNA-Mediated Laccase Silencing. Plant Cell 2018, 30, 2649–2662. [Google Scholar] [CrossRef]
- Kaur, R.; Bhunia, R.K.; Rajam, M.V. MicroRNAs as potential targets for improving rice yield via plant architecture modulation: Recent studies and future perspectives. J. Biosci. 2020, 45, 116. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Wang, Z.; Li, X.; Sun, H. Evidence of the Regulatory Roles of Candidate miRNAs During Somatic Embryogenesis in Lilium davidii var. unicolor. J. Plant Growth Regul. 2020, 40, 197–214. [Google Scholar] [CrossRef]
- Lu, S.; Li, Q.; Wei, H.; Chang, M.J.; Tunlaya-Anukit, S.; Kim, H.; Liu, J.; Song, J.; Sun, Y.-H.; Yuan, L.; et al. Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. USA 2013, 110, 10848–10853. [Google Scholar] [CrossRef] [PubMed]
- Souer, E.; van Houwelingen, A.; Kloos, D.; Mol, J.; Koes, R. The No Apical Meristem Gene of Petunia Is Required for Pattern Formation in Embryos and Flowers and Is Expressed at Meristem and Primordia Boundaries. Cell 1996, 85, 159–170. [Google Scholar] [CrossRef]
- Fang, Y.-N.; Yang, X.-M.; Jiang, N.; Wu, X.-M.; Guo, W.-W. Genome-wide identification and expression profiles of phased siRNAs in a male-sterile somatic cybrid of pummelo (Citrus grandis). Tree Genet. Genomes 2020, 16, 46. [Google Scholar] [CrossRef]
- Fang, Y.-N.; Qiu, W.-M.; Wang, Y.; Wu, X.-M.; Xu, Q.; Guo, W.-W. Identification of differentially expressed microRNAs from a male sterile Ponkan mandarin (Citrus reticulata Blanco) and its fertile wild type by small RNA and degradome sequencing. Tree Genet. Genomes 2014, 10, 1567–1581. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Ai, X.Y.; Guo, W.W.; Peng, S.A.; Deng, X.X.; Hu, C.G. Identification of miRNAs and their target genes using deep sequencing and degradome analysis in trifoliate orange [Poncirus trifoliata L. Raf] [corrected]. Mol. Biotechnol. 2012, 51, 44–57. [Google Scholar] [CrossRef]
- Gao, Y.; Wei, W.; Zhao, X.; Tan, X.; Fan, Z.; Zhang, Y.; Jing, Y.; Meng, L.; Zhu, B.; Zhu, H.; et al. A NAC transcription factor, NOR-like1, is a new positive regulator of tomato fruit ripening. Hortic. Res. 2018, 5, 75. [Google Scholar] [CrossRef]
- Gong, X.; Zhao, L.; Song, X.; Lin, Z.; Gu, B.; Yan, J.; Zhang, S.; Tao, S.; Huang, X. Genome-wide analyses and expression patterns under abiotic stress of NAC transcription factors in white pear (Pyrus bretschneideri). BMC Plant Biol. 2019, 19, 161. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway | Gene Numbers | |||
|---|---|---|---|---|
| 100 DAF | 130 DAF | 175 DAF | 205 DAF | |
| Metabolic pathways | 252 | 301 | 273 | 237 |
| Biosynthesis of secondary metabolites | 169 | 191 | 157 | 157 |
| Plant–pathogen interaction | 48 | 34 | 46 | 30 |
| Plant hormone signal transduction | 41 | 25 | 43 | 29 |
| Protein processing in endoplasmic reticulum | 34 | 32 | 15 | 17 |
| Starch and sucrose metabolism | 34 | 28 | 32 | 29 |
| Phenylpropanoid biosynthesis | 31 | 32 | 25 | 35 |
| Biosynthesis of amino acids | 31 | 34 | 35 | 24 |
| Carbon metabolism | 31 | 44 | 40 | 18 |
| Glycolysis/Gluconeogenesis | 17 | 26 | 15 | 14 |
| Terpenoid backbone biosynthesis | 17 | 14 | 15 | 10 |
| Sesquiterpenoid and triterpenoid biosynthesis | 15 | 19 | 16 | 12 |
| Peroxisome | 14 | 15 | 15 | 13 |
| Fatty acid metabolism | 13 | 11 | 12 | 14 |
| Pentose and glucuronate interconversions | 13 | 15 | 15 | 11 |
| Fatty acid degradation | 11 | 15 | 9 | 12 |
| Glyoxylate and dicarboxylate metabolism | 10 | 19 | 17 | 9 |
| Ascorbate and aldarate metabolism | 10 | 11 | 6 | 5 |
| Biosynthesis of unsaturated fatty acids | 9 | 5 | 6 | 6 |
| Carotenoid biosynthesis | 9 | 8 | 8 | 6 |
| Tropane, piperidine and pyridine alkaloid biosynthesis | 9 | 6 | 5 | 7 |
| Ubiquitin mediated proteolysis | 9 | 12 | 7 | 9 |
| Photosynthesis | 8 | 35 | 25 | 12 |
| Nitrogen metabolism | 8 | 10 | 8 | 7 |
| Fatty acid biosynthesis | 7 | 7 | 9 | 11 |
| Degradation of aromatic compounds | 6 | 8 | 5 | 5 |
| Cutin, suberine and wax biosynthesis | 6 | 8 | 6 | 9 |
| Flavonoid biosynthesis | 5 | 12 | 9 | 25 |
| Gene Id | Gene Description | Log2ratio (MT/WT) | |||
|---|---|---|---|---|---|
| 100 DAF | 130 DAF | 175 DAF | 205 DAF | ||
| Starch and sucrose metabolism | |||||
| Cs7g16980 | GAUT | −1.71 | −1.37 | - | −1.17 |
| Cs6g10710 | GAUT | −0.98 | - | - | - |
| Cs4g06850 | SS | −1.11 | - | - | −1.16 |
| Cs6g14340 | β-fructofuranosidase | −3.60 | −3.04 | - | −3.12 |
| Cs5g09220 | β-fructofuranosidase | 1.65 | 2.66 | - | - |
| Cs7g05690 | SPS | 1.53 | 2.57 | 1.69 | 1.96 |
| orange1.1t03668 | SPS | 1.09 | 0.66 | 0.75 | 0.67 |
| Cs4g05380 | SPS | 0.96 | 1.04 | - | - |
| Cs5g19060 | SPS | 1.57 | - | −2.90 | −1.88 |
| Cs2g10900 | α-glucosidase | −0.77 | - | 3.04 | 1.86 |
| Cs4g06710 | PE | −1.07 | −1.82 | −1.29 | −1.55 |
| Cs5g33420 | PE | −1.84 | - | 3.97 | - |
| Cs2g17070 | PE | 1.71 | - | - | −2.97 |
| orange1.1t02530 | PG | - | - | - | 6.66 |
| orange1.1t02529 | PG | - | - | - | 6.22 |
| Carotenoid biosynthesis | |||||
| orange1.1t02108 | PSY | −1.69 | - | −1.77 | −3.48 |
| Cs6g15910 | PSY | - | −0.84 | −1.16 | - |
| Cs3g11040 | ZDS | 1.17 | - | - | - |
| Cs4g14850 | LCYE | 0.83 | - | - | - |
| Cs9g19270 | CHYB | 1.05 | - | - | - |
| Cs5g14370 | NCED | −1.63 | −1.86 | −2.07 | - |
| Cs9g11260 | NCED | - | - | −4.52 | - |
| Cs2g03270 | NCED | - | - | - | 2.58 |
| Plant hormone signal transduction | |||||
| Cs5g34270 | SnRK2 | −0.69 | - | - | - |
| Cs5g07700 | SnRK2 | −1.80 | - | - | - |
| Cs1g23060 | SnRK2 | −0.78 | −1.47 | - | −1.51 |
| Cs8g19140 | PP2C | 1.09 | - | - | - |
| Cs4g14980 | PP2C | −1.28 | - | - | - |
| Cs5g03060 | ACS | - | −1.50 | −0.93 | - |
| orange1.1t00416 | ACS | −3.32 | - | −2.86 | - |
| Cs2g02500 | ACO | 2.31 | - | - | - |
| Cs5g20590 | ACO | - | −0.91 | - | - |
| Cs9g08850 | ETR | −1.70 | - | −0.84 | - |
| orange1.1t02185 | MPK | 0.77 | 1.34 | 1.28 | 1.35 |
| Cs4g17960 | EBF1 | −2.08 | - | - | - |
| Cs5g29870 | ERF1 | −3.32 | - | −4.13 | - |
| Cs9g10650 | ERF1 | −4.24 | - | −5.21 | −3.53 |
| orange1.1t01539 | AHP | 1.90 | 3.58 | 1.74 | 1.05 |
| Cs9g02730 | B-ARR | 0.69 | - | - | - |
| Cs4g01640 | B-ARR | 0.84 | - | - | - |
| Cs8g02900 | AUX1 | 2.26 | - | - | −7.32 |
| Cs7g31320 | AUX1 | 1.32 | - | - | - |
| orange1.1t00464 | JAR1 | −1.42 | −0.95 | −1.35 | - |
| Cs1g17220 | JAZ | −1.15 | - | −1.28 | - |
| Cs4g06520 | JAZ | −0.82 | - | −0.81 | - |
| Cs7g02820 | JAZ | −2.21 | - | - | - |
| Cs4g07130 | JAZ | −0.91 | - | −1.14 | - |
| Cs1g17210 | JAZ | −1.53 | - | −1.00 | - |
| orange1.1t00550 | MYC2 | −2.37 | - | −2.00 | - |
| orange1.1t01021 | MYC2 | −0.85 | −0.83 | −1.23 | - |
| Cs5g01450 | MYC2 | −0.94 | - | −0.93 | - |
| Cs7g19070 | TGA | 7.07 | 3.70 | - | −2.26 |
| orange1.1t03769 | LOX | 6.68 | - | - | - |
| orange1.1t04376 | LOX | −2.12 | −1.13 | −0.92 | - |
| Cs1g17380 | LOX | −1.14 | −0.90 | −1.06 | - |
| orange1.1t03771 | LOX | −7.34 | - | - | - |
| Cs3g24230 | AOS | −1.46 | - | - | - |
| orange1.1t03727 | OPR | −1.33 | - | - | - |
| Cs5g17900 | OPR | −1.47 | - | −2.74 | - |
| Cs4g15220 | OPR | −1.02 | - | - | 0.77 |
| Cs5g17880 | OPR | −1.21 | - | −2.73 | - |
| Sample | Clean Reads | Total Reads (18–30 nt) | Unique Reads | Mapped sRNA | Known-miRNA | Novel-miRNA |
|---|---|---|---|---|---|---|
| M_100D | 14,316,231 | 4,176,010 | 1,518,461 | 3,246,961 (77.75%) | 65,163 (2.01%) | 3554 (0.11%) |
| M_130D | 15,465,813 | 5,296,231 | 1,879,119 | 4,146,086 (78.28%) | 114,988 (2.77%) | 5976 (0.14%) |
| M_175D | 16,418,922 | 1,906,354 | 818,850 | 1,385,348 (72.67%) | 29,872 (2.16%) | 1019 (0.07%) |
| M_205D | 15,237,868 | 8,968,818 | 2,202,926 | 6,648,319 (74.13%) | 196,588 (2.96%) | 11,140 (0.17%) |
| W_100D | 16,423,940 | 8,762,671 | 2,381,531 | 6,451,132 (73.62%) | 236,285 (3.66%) | 11,183 (0.17%) |
| W_130D | 16,986,556 | 6,362,575 | 1,736,884 | 5,148,648 (80.92%) | 164,510 (3.20%) | 7221 (0.14%) |
| W_175D | 14,755,090 | 3,676,819 | 1,068,268 | 2,970,459 (80.79%) | 39,118 (1.32%) | 4677 (0.16%) |
| W_205D | 16,507,044 | 7,430,258 | 1,729,613 | 5,695,338 (76.65% | 225,001 (3.95%) | 12,835 (0.23%) |
| miRNA/Target Gene | Target-Start | Target-End | Score | log2 (MT/WT) | Target Annotation | |||
|---|---|---|---|---|---|---|---|---|
| 100 DAF | 130 DAF | 175 DAF | 205 DAF | |||||
| csi-miR156f-5p | −3.48 | −1.11 | 3.55 | −1.25 | ||||
| orange1.1t02597 | 1203 | 1223 | 0.5 | SP13B | ||||
| Cs7g10990 | 1536 | 1556 | 0.5 | SPL16 | ||||
| Cs1g03640 | 1585 | 1605 | 0.5 | SPL7 | ||||
| Cs2g05730 | 807 | 827 | 1.5 | SPB1 | ||||
| orange1.1t02265 | 996 | 1016 | 1.5 | SPL9 | ||||
| Cs2g23550 | 892 | 912 | 1.5 | SPB2 | ||||
| Cs7g11770 | 1477 | 1497 | 1.5 | SPL6 | ||||
| Cs7g10830 | 1172 | 1192 | 1.5 | SPL2 | ||||
| csi-miR160c-5p | 1.43 | 1.24 | −1.94 | −3.09 | ||||
| Cs8g16440 | 1967 | 1987 | 0.5 | ARFR | ||||
| Cs7g25670 | 1730 | 1750 | 0.5 | ARFR | ||||
| Cs6g11800 | 2142 | 2162 | 2 | ARFR | ||||
| Cs3g18940 | 1412 | 1432 | 2 | ARFQ | ||||
| csi-miR397-5p | 1.61 | – | 7.88 | 3.8 | ||||
| Cs6g11860 | 736 | 756 | 1.5 | LAC11 | ||||
| Cs6g07800 | 734 | 754 | 1.5 | LAC4 | ||||
| Cs8g19850 | 669 | 689 | 2 | LAC4 | ||||
| Cs6g07410 | 764 | 784 | 2 | LAC7 | ||||
| Cs7g23490 | 705 | 725 | 2.5 | LAC17 | ||||
| Cs6g07400 | 789 | 809 | 2.5 | LAC7 | ||||
| Cs6g07450 | 878 | 898 | 2.5 | LAC7 | ||||
| Cs8g18800 | 711 | 731 | 2.5 | LAC17 | ||||
| Cs6g06920 | 693 | 713 | 3 | LAC17 | ||||
| Cs6g06880 | 713 | 733 | 3 | LAC17 | ||||
| Cs8g17630 | 724 | 744 | 3 | LAC17 | ||||
| Cs8g17350 | 690 | 710 | 3.5 | LAC17 | ||||
| Cs6g06890 | 789 | 809 | 3.5 | LAC17 | ||||
| Cs7g31620 | 685 | 705 | 3.5 | LAC22 | ||||
| csi-miR172a-3p | – | −2.69 | 7.3 | – | ||||
| Cs7g27790 | 1604 | 1623 | 1 | RAP27 | ||||
| Cs6g04120 | 1535 | 1554 | 2 | AP2 | ||||
| orange1.1t04055 | 2039 | 2058 | 2 | AP2 | ||||
| Cs8g17390 | 1810 | 1829 | 2 | RAP27 | ||||
| csi-miR3954 | – | – | 1.25 | 1.04 | ||||
| orange1.1t05093 | 75 | 95 | 2 | NAC40 | ||||
| Cs1g09660 | 148 | 168 | 2 | NAC40 | ||||
| orange1.1t05587 | 237 | 257 | 2.5 | NAC79 | ||||
| Cs1g09710 | 156 | 176 | 3 | NAC40 | ||||
| Cs7g22510 | 75 | 95 | 4 | NAC4 | ||||
| Cs7g22470 | 178 | 198 | 4 | NAC4 | ||||
| Cs7g22540 | 75 | 95 | 4 | NAC91 | ||||
| miRN106 | −5.76 | −7.61 | −6.33 | −2.79 | ||||
| Cs2g08080 | 2017 | 2037 | 3.5 | TCP2 | ||||
| Cs3g06390 | 762 | 782 | 4 | MYB33 | ||||
| Cs8g16870 | 204 | 224 | 4.5 | NAC29 | ||||
| Cs2g15820 | 1428 | 1448 | 4.5 | TCP4 | ||||
| Cs7g25460 | 2050 | 2070 | 4.5 | TCP4 | ||||
| miRN127 | −2.16 | −4.45 | −8.91 | −6.15 | ||||
| orange1.1t03355 | 751 | 773 | 5 | PP323 | ||||
| miRN54 | – | – | −8.65 | −1.05 | ||||
| Cs1g19320 | 1097 | 1120 | 5 | HSR4 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mi, L.; Ma, D.; Lv, S.; Xu, S.; Zhong, B.; Peng, T.; Liu, D.; Liu, Y. Comparative Transcriptome and sRNAome Analyses Reveal the Regulatory Mechanisms of Fruit Ripening in a Spontaneous Early-Ripening Navel Orange Mutant and Its Wild Type. Genes 2022, 13, 1706. https://doi.org/10.3390/genes13101706
Mi L, Ma D, Lv S, Xu S, Zhong B, Peng T, Liu D, Liu Y. Comparative Transcriptome and sRNAome Analyses Reveal the Regulatory Mechanisms of Fruit Ripening in a Spontaneous Early-Ripening Navel Orange Mutant and Its Wild Type. Genes. 2022; 13(10):1706. https://doi.org/10.3390/genes13101706
Chicago/Turabian StyleMi, Lanfang, Dong Ma, Shuping Lv, Saibing Xu, Balian Zhong, Ting Peng, Dechun Liu, and Yong Liu. 2022. "Comparative Transcriptome and sRNAome Analyses Reveal the Regulatory Mechanisms of Fruit Ripening in a Spontaneous Early-Ripening Navel Orange Mutant and Its Wild Type" Genes 13, no. 10: 1706. https://doi.org/10.3390/genes13101706
APA StyleMi, L., Ma, D., Lv, S., Xu, S., Zhong, B., Peng, T., Liu, D., & Liu, Y. (2022). Comparative Transcriptome and sRNAome Analyses Reveal the Regulatory Mechanisms of Fruit Ripening in a Spontaneous Early-Ripening Navel Orange Mutant and Its Wild Type. Genes, 13(10), 1706. https://doi.org/10.3390/genes13101706