
BRCA2 Haploinsufficiency in Telomere Maintenance


Abstract
:1. Introduction
2. Materials and Methods
2.1. Lymphoid Cell Lines
2.2. Chromosomal Analysis
2.3. Statistical Analysis
3. Results
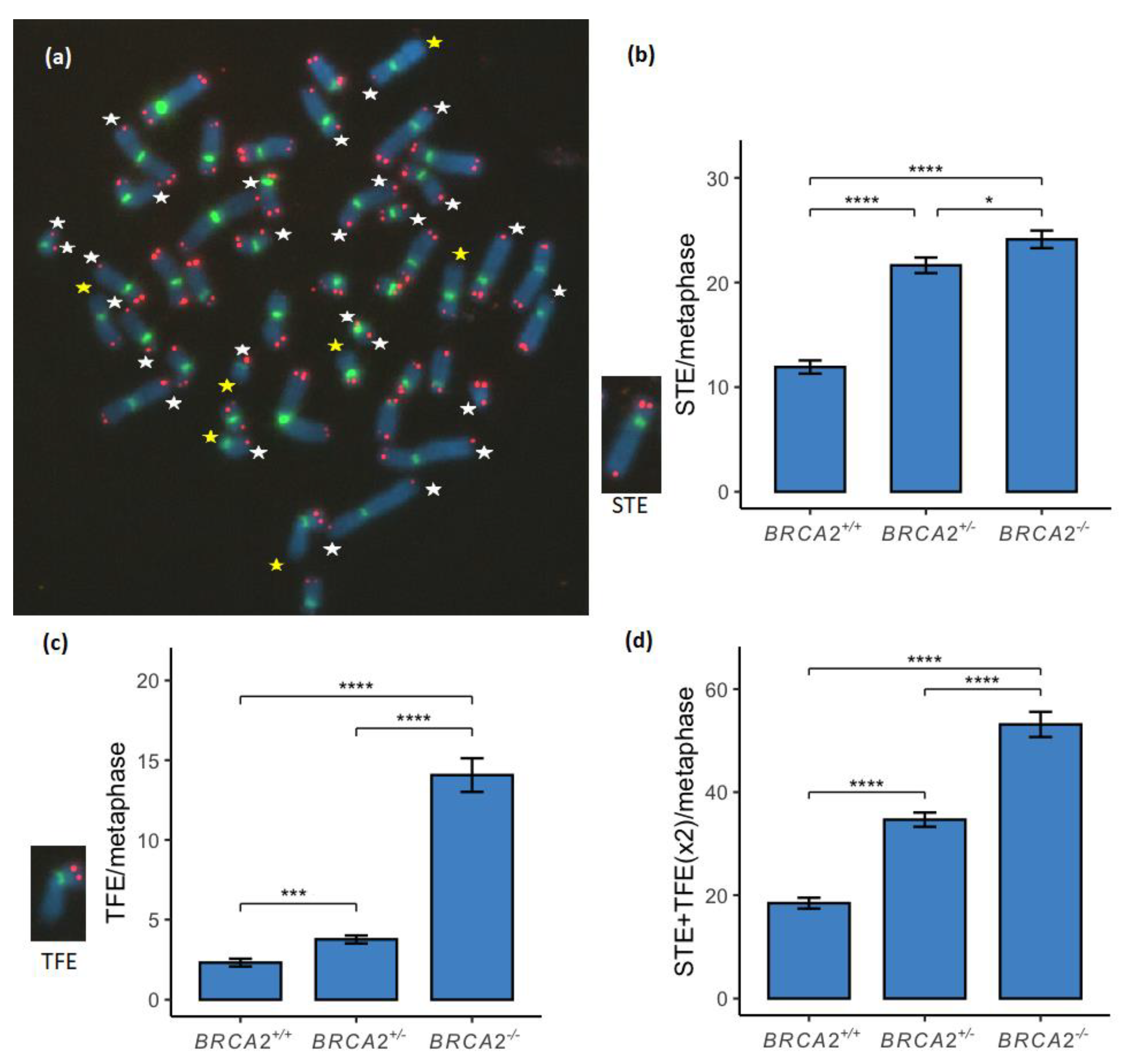
3.1. Telomere Loss Was Found to Increase in a Stepwise Manner between the BRCA2 Genotypes
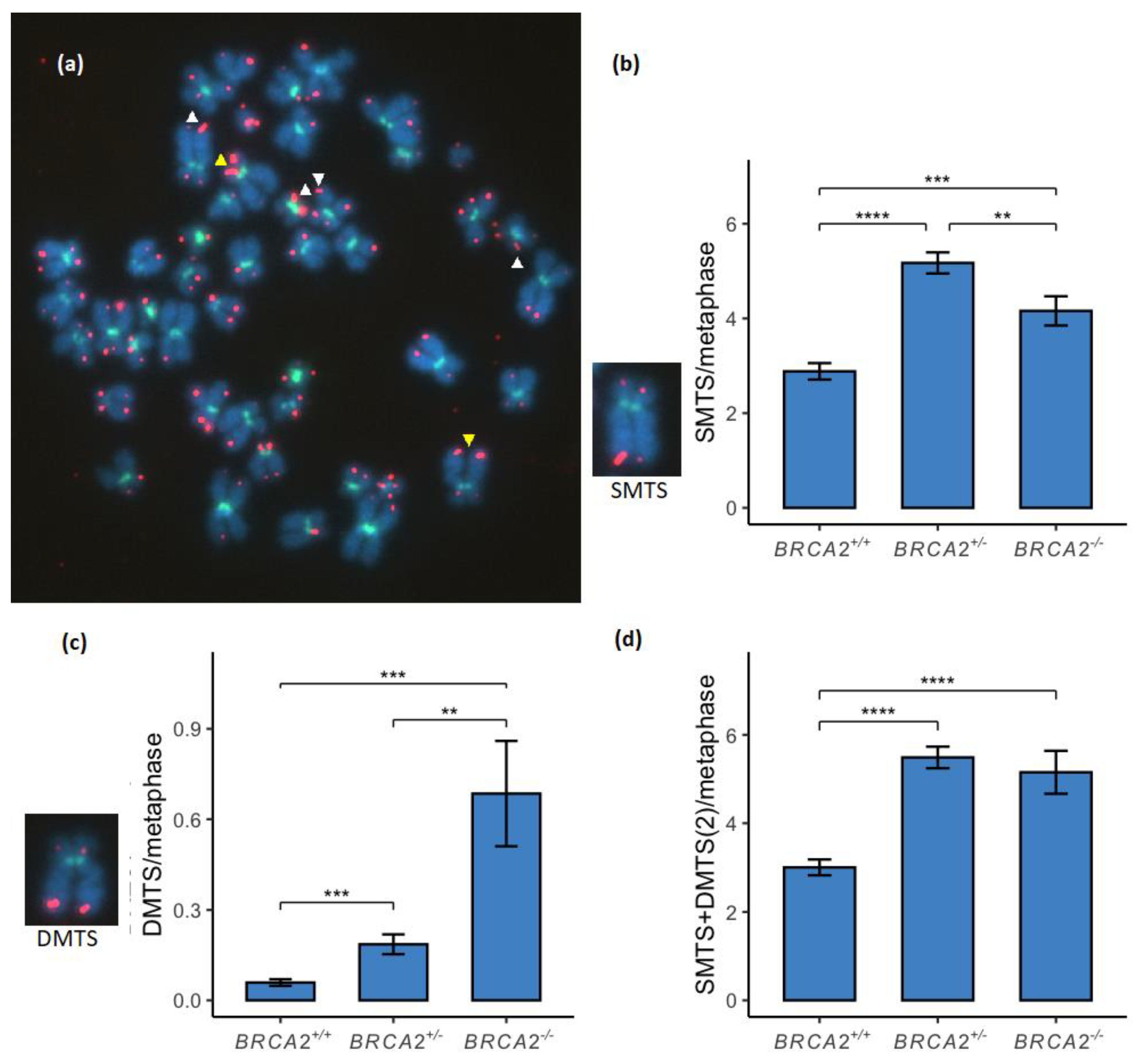
3.2. Multitelomeric Signals Increase between BRCA2 Wild-Type and Mutated Genotypes
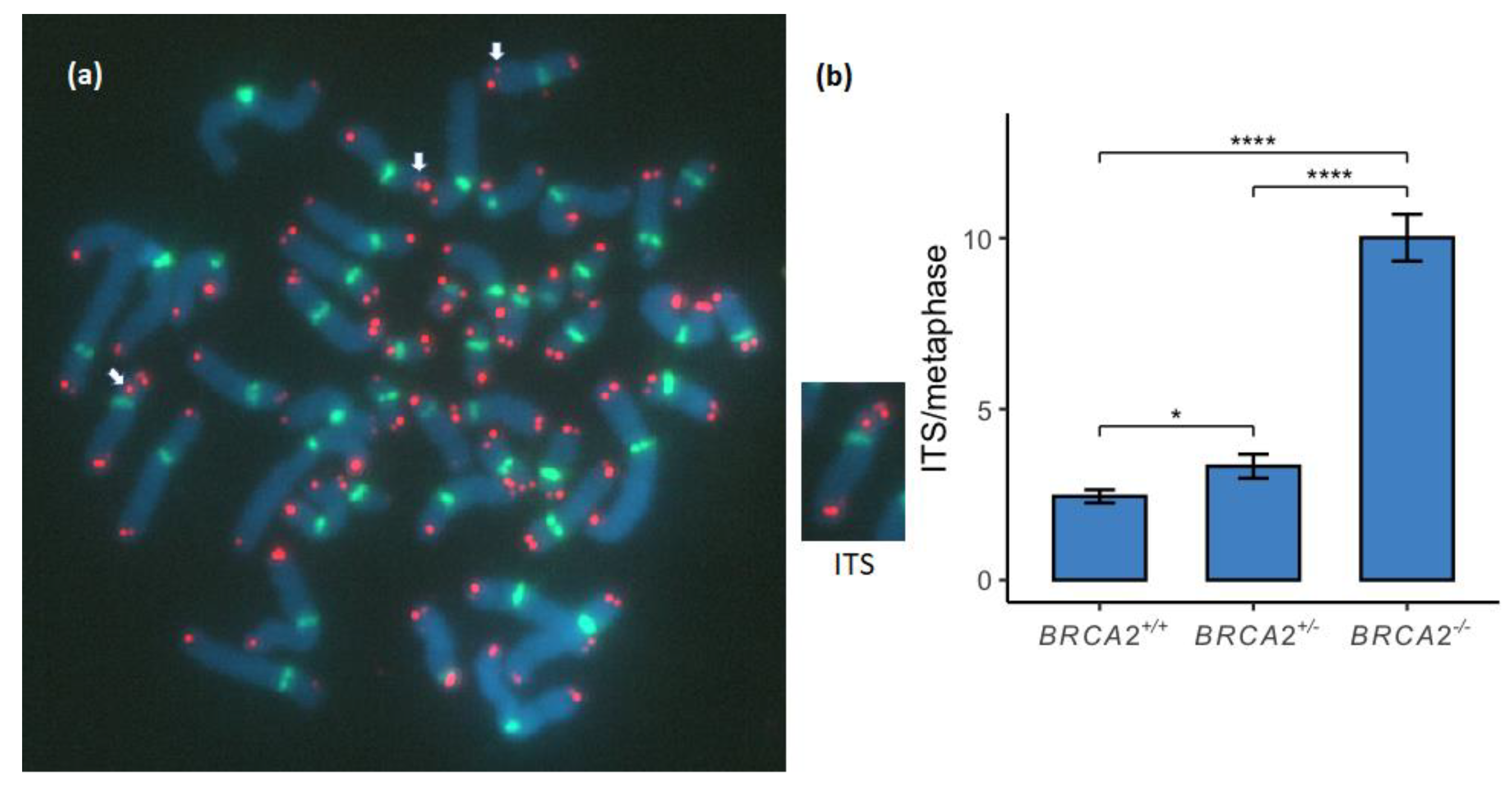
3.3. Interstitial Telomere Sequences Increase in a Stepwise Manner between the BRCA2 Genotypes
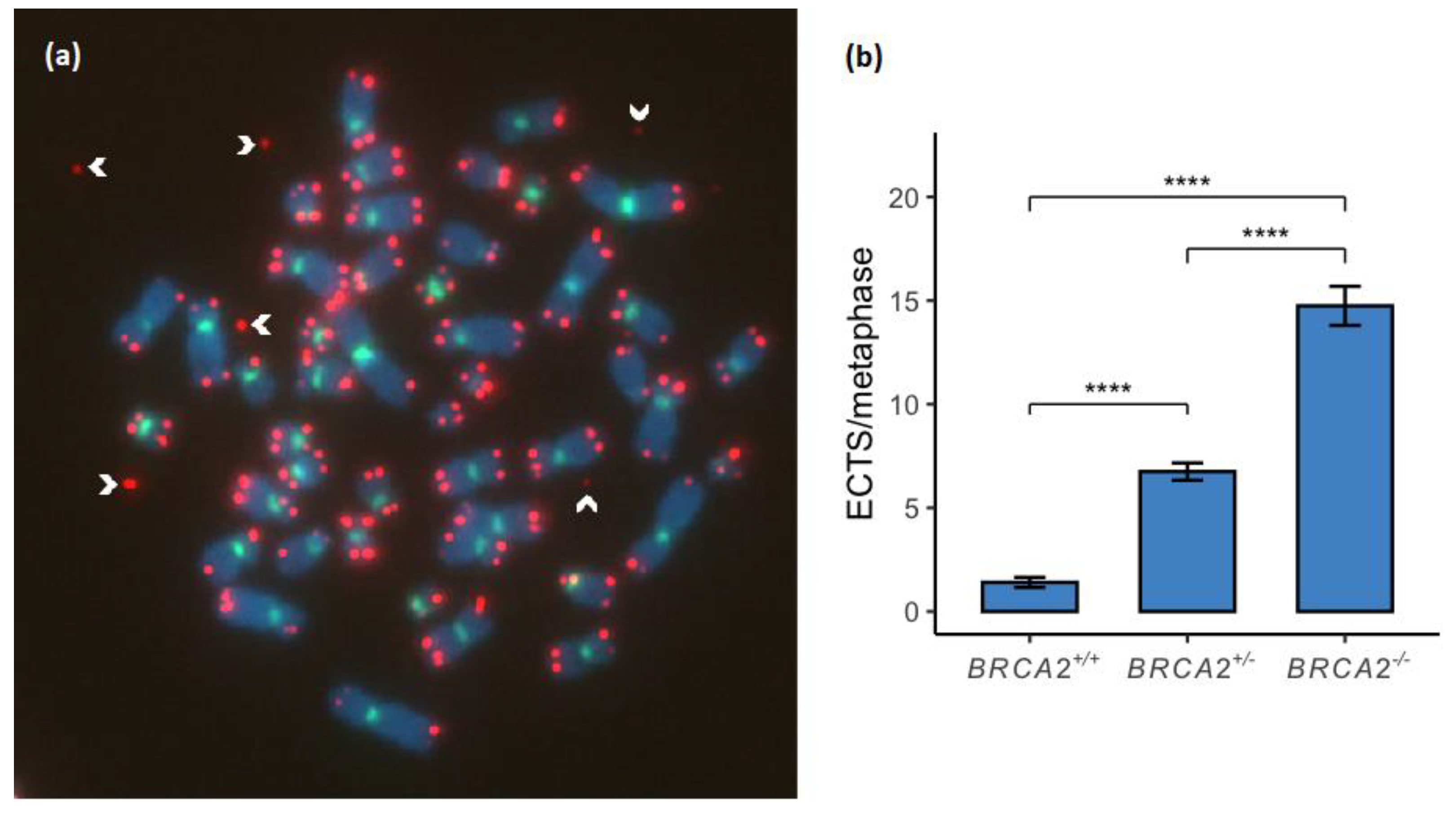
3.4. Extrachromosomal Telomere Sequences Increase in a Stepwise Manner between the BRCA2 Genotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fradet-Turcotte, A.; Sitz, J.; Grapton, D.; Orthwein, A. BRCA2 functions: From DNA repair to replication fork stabilization. Endocr. Relat. Cancer 2016, 23, T1–T17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, H.P.; Heyer, W.-D.; Liu, J. Guardians of the Genome: BRCA2 and Its Partners. Genes 2021, 12, 1229. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius, S.; Olafsdottir, G.; Tryggvadottir, L.; Neuhausen, S.; Jonasson, J.G.; Tavtigian, S.V.; Tulinius, H.; Ögmundsdottir, H.M.; Eyfjörd, J.E. A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nat. Genet. 1996, 13, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Tulinius, H.; Olafsdottir, G.H.; Sigvaldason, H.; Arason, A.; Barkardottir, R.B.; Egilsson, V.; Ogmundsdottir, H.M.; Tryggvadottir, L.; Gudlaugsdottir, S.; Eyfjord, J.E. The effect of a single BRCA2 mutation on cancer in Iceland. J. Med. Genet. 2002, 39, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Rafnar, T.; Sigurjonsdottir, G.R.; Stacey, S.N.; Halldorsson, G.; Sulem, P.; Pardo, L.M.; Helgason, H.; Sigurdsson, S.T.; Gudjonsson, T.; Tryggvadottir, L.; et al. Association of BRCA2 K3326* With Small Cell Lung Cancer and Squamous Cell Cancer of the Skin. J. Natl. Cancer Inst. 2018, 110, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Dullens, B.; de Putter, R.; Lambertini, M.; Toss, A.; Han, S.; Van Nieuwenhuysen, E.; Van Gorp, T.; Vanderstichele, A.; Van Ongeval, C.; Keupers, M.; et al. Cancer Surveillance in Healthy Carriers of Germline Pathogenic Variants in BRCA1/2: A Review of Secondary Prevention Guidelines. J. Oncol. 2020, 2020, 9873954. [Google Scholar] [CrossRef]
- Thorlacius, S.; Sigurdsson, S.; Bjarnadottir, H.; Olafsdottir, G.; Jonasson, J.G.; Tryggvadottir, L.; Tulinius, H.; Eyfjörd, J.E. Study of a single BRCA2 mutation with high carrier frequency in a small population. Am. J. Hum. Genet. 1997, 60, 1079–1084. [Google Scholar]
- Mikaelsdottir, E.K.; Valgeirsdottir, S.; Eyfjord, J.E.; Rafnar, T. The Icelandic founder mutation BRCA2 999del5: Analysis of expression. Breast Cancer Res. 2004, 6, R284–R290. [Google Scholar] [CrossRef] [Green Version]
- Bodvarsdottir, S.K.; Steinarsdottir, M.; Bjarnason, H.; Eyfjord, J.E. Dysfunctional telomeres in human BRCA2 mutated breast tumors and cell lines. Mutat. Res. 2012, 729, 90–99. [Google Scholar] [CrossRef]
- Gretarsdottir, S.; Thorlacius, S.; Valgardsdottir, R.; Gudlaugsdottir, S.; Sigurdsson, S.; Steinarsdottir, M.; Jonasson, J.G.; Anamthawat-Jonsson, K.; Eyfjörd, J.E. BRCA2 and p53 mutations in primary breast cancer in relation to genetic instability. Cancer Res. 1998, 58, 859–862. [Google Scholar]
- Patel, K.J.; Yu, V.P.; Lee, H.; Corcoran, A.; Thistlethwaite, F.C.; Evans, M.J.; Colledge, W.H.; Friedman, L.S.; Ponder, B.A.; Venkitaraman, A.R. Involvement of Brca2 in DNA Repair. Mol. Cell 1998, 1, 347–357. [Google Scholar] [CrossRef]
- Eyfjord, J.E.; Bodvarsdottir, S.K. Genomic instability and cancer: Networks involved in response to DNA damage. Mutat. Res. 2005, 592, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic Inactivation of BRCA2 in Fanconi Anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Alter, B.P.; Rosenberg, P.S.; Brody, L.C. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J. Med. Genet. 2007, 44, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Badie, S.; Escandell, J.M.; Bouwman, P.; Carlos, A.R.; Thanasoula, M.; Gallardo, M.M.; Suram, A.; Jaco, I.; Benitez, J.; Herbig, U.; et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 2010, 17, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Choi, E.S.; Hwang, K.; Kim, J.; Sampath, S.; Venkitaraman, A.R.; Lee, H. The Breast Cancer Susceptibility Gene BRCA2 Is Required for the Maintenance of Telomere Homeostasis. J. Biol. Chem. 2012, 287, 5091–5101. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, B.; Aradottir, M.; Stefansson, O.A.; Bodvarsdottir, S.K.; Eyfjörd, J.E. Telomere length is predictive of breast cancer risk in BRCA2 mutation carriers. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1248–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudson, A.G., Jr. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefansson, O.A.; Jonasson, J.G.; Olafsdottir, K.; Bjarnason, H.; Th Johannsson, O.; Bodvarsdottir, S.K.; Valgeirsdottir, S.; Eyfjord, J.E. Genomic and phenotypic analysis of BRCA2mutated breast cancers reveals co-occurring changes linked to progression. Breast Cancer Res. 2011, 13, R95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aradottir, M.; Reynisdottir, S.T.; Stefansson, O.A.; Jonasson, J.G.; Sverrisdottir, A.; Tryggvadottir, L.; Eyfjord, J.E.; Bodvarsdottir, S.K. Aurora A is a prognostic marker for breast cancer arising inBRCA2mutation carriers. J. Pathol. Clin. Res. 2015, 1, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Cassidy, L.D.; Pisupati, V.; Jonasson, J.G.; Bjarnason, H.; Eyfjord, J.E.; Karreth, F.A.; Lim, M.; Barber, L.M.; Clatworthy, S.A.; et al. Germline Brca2 Heterozygosity Promotes KrasG12D -Driven Carcinogenesis in a Murine Model of Familial Pancreatic Cancer. Cancer Cell 2010, 18, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Karaayvaz-Yildirim, M.; Silberman, R.E.; Langenbucher, A.; Saladi, S.V.; Ross, K.N.; Zarcaro, E.; Desmond, A.; Yildirim, M.; Vivekanandan, V.; Ravichandran, H.; et al. Aneuploidy and a deregulated DNA damage response suggest haploinsufficiency in breast tissues of BRCA2 mutation carriers. Sci. Adv. 2020, 6, eaay2611. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.S.; Tarsounas, M. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.S.; Lee, J.J.; Min, J.; Hwang, K.; Park, S.G.; Kim, E.H.; Kim, B.C.; Bhak, J.; Lee, H. Brca2 abrogation engages with the alternative lengthening of telomeres via break-induced replication. FEBS J. 2019, 286, 1841–1858. [Google Scholar] [CrossRef]
- Min, K.B.; Min, J.Y. Association between leukocyte telomere length and serum carotenoid in US adults. Eur. J. Nutr. 2017, 56, 1045–1052. [Google Scholar] [CrossRef]
- Ozer, O.; Bhowmick, R.; Liu, Y.; Hickson, I.D. Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 2018, 9, 15836–15846. [Google Scholar] [CrossRef] [PubMed]
- Popp, H.; Kalb, R.; Fischer, A.; Lobitz, S.; Kokemohr, I.; Hanenberg, H.; Schindler, D. Screening Fanconi anemia lymphoid cell lines of non-A, C, D2, E, F, G subtypes for defects in BRCA2/FANCD1. Cytogenet. Genome Res. 2003, 103, 54–57. [Google Scholar] [CrossRef]
- Castells-Roca, L.; Gutiérrez-Enríquez, S.; Bonache, S.; Bogliolo, M.; Carrasco, E.; Aza-Carmona, M.; Montalban, G.; Muñoz-Subirana, N.; Pujol, R.; Cruz, C.; et al. Clinical consequences of BRCA2 hypomorphism. NPJ Breast Cancer 2021, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Cherdyntseva, V.; Gagos, S. Chromosome extremities under the microscopy lens: Molecular cytogenetics in telomere research. Curr. Opin. Genet. Dev. 2020, 60, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Williams, E.S.; Cornforth, M.N.; Goodwin, E.H. Chromosome Orientation Fluorescence In Situ Hybridization or Strand-Specific FISH. Methods Mol. Biol. 2010, 659, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell. Biol. 2017, 37, e00226-17. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, J.; Tacconi, E.M.C.; Folio, C.; Badie, S.; Porru, M.; Klare, K.; Tumiati, M.; Markkanen, E.; Halder, S.; Ryan, A.; et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell. 2016, 61, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Aksenova, A.Y.; Mirkin, S.M. At the Beginning of the End and in the Middle of the Beginning: Structure and Maintenance of Telomeric DNA Repeats and Interstitial Telomeric Sequences. Genes 2019, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef]
- Pompili, L.; Maresca, C.; Dello Stritto, A.; Biroccio, A.; Salvati, E. BRCA2 Deletion Induces Alternative Lengthening of Telomeres in Telomerase Positive Colon Cancer Cells. Genes 2019, 10, 697. [Google Scholar] [CrossRef] [Green Version]
- Roumelioti, F.M.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D.; Gagos, S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal-Garcia, J.; Crown, K.N.; Ramsden, D.A.; Sekelsky, J. DNA polymerase theta suppresses mitotic crossing over. PLoS Genet. 2021, 17, e1009267. [Google Scholar] [CrossRef] [PubMed]
- Hengel, S.R.; Spies, M.A.; Spies, M. Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell Chem. Biol. 2017, 24, 1101–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.S.; Pomerantz, R.T. DNA Polymerase theta: A Cancer Drug Target with Reverse Transcriptase Activity. Genes 2021, 12, 1146. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Poltheta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Kelso, A.A.; Lopezcolorado, F.W.; Bhargava, R.; Stark, J.M. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. 2019, 15, e1008319. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.S.; Algouneh, A.; Hakem, R. Exploiting synthetic lethality to target BRCA1/2-deficient tumors: Where we stand. Oncogene 2021, 40, 3001–3014. [Google Scholar] [CrossRef]
- Jonasson, J.G.; Stefansson, O.A.; Johannsson, O.T.; Sigurdsson, H.; Agnarsson, B.A.; Olafsdottir, G.H.; Alexiusdottir, K.K.; Stefansdottir, H.; Mitev, R.M.; Olafsdottir, K.; et al. Oestrogen receptor status, treatment and breast cancer prognosis in Icelandic BRCA2 mutation carriers. Br. J. Cancer 2016, 115, 776–783. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lymphoid Cell Lines | BRCA2 Genotype 1 | BRCA2 Mutation(s) | |
|---|---|---|---|
| EB0392 | +/+ | None | |
| EB6457 | +/+ | None | |
| EB1482 | +/- | 999del5 2 | |
| EB1690 | +/- | 999del5 | |
| EB1830 | +/- | 999del5 | |
| EB2302 | +/- | 999del5 | |
| EB6085 | +/- | 999del5 | |
| HSC62N 3 | -/- | IVS 19-1 G > A | IVS 19-1 G > A |
| NORD 4 | -/- | 886delGT | 8447T > A |
| SPAN 5 | -/- | 15-16 exons del | 1597del |
| Lymphoid Cell Lines | BRCA2 Genotype | Metaphases Analyzed | Mean Chromosome Number | Chromosome Number Range |
|---|---|---|---|---|
| EB0392 | +/+ | 61 | 46.5 | 31–92 |
| EB6457 | +/+ | 65 | 45.4 | 31–83 |
| EB1482 | +/- | 64 | 48.0 | 38–92 |
| EB1690 | +/- | 60 | 47.6 | 41–92 |
| EB1830 | +/- | 63 | 47.6 | 26–92 |
| EB2302 | +/- | 62 | 46.6 | 45–92 |
| EB6085 | +/- | 60 | 46.6 | 44–93 |
| HSC62N | -/- | 43 | 46.3 | 36–87 |
| NORD | -/- | 63 | 44.7 | 35–46 |
| SPAN | -/- | 66 | 45.0 | 39–47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunnarsdottir, S.R.; Bjarnason, H.; Thorvaldsdottir, B.; Paland, F.; Steinarsdottir, M.; Eyfjörd, J.E.; Bödvarsdottir, S.K. BRCA2 Haploinsufficiency in Telomere Maintenance. Genes 2022, 13, 83. https://doi.org/10.3390/genes13010083
Gunnarsdottir SR, Bjarnason H, Thorvaldsdottir B, Paland F, Steinarsdottir M, Eyfjörd JE, Bödvarsdottir SK. BRCA2 Haploinsufficiency in Telomere Maintenance. Genes. 2022; 13(1):83. https://doi.org/10.3390/genes13010083
Chicago/Turabian StyleGunnarsdottir, Soffía R., Hördur Bjarnason, Birna Thorvaldsdottir, Felice Paland, Margrét Steinarsdottir, Jórunn E. Eyfjörd, and Sigrídur K. Bödvarsdottir. 2022. "BRCA2 Haploinsufficiency in Telomere Maintenance" Genes 13, no. 1: 83. https://doi.org/10.3390/genes13010083
APA StyleGunnarsdottir, S. R., Bjarnason, H., Thorvaldsdottir, B., Paland, F., Steinarsdottir, M., Eyfjörd, J. E., & Bödvarsdottir, S. K. (2022). BRCA2 Haploinsufficiency in Telomere Maintenance. Genes, 13(1), 83. https://doi.org/10.3390/genes13010083