
Fanconi Anaemia, Childhood Cancer and the BRCA Genes


Abstract
1. Introduction
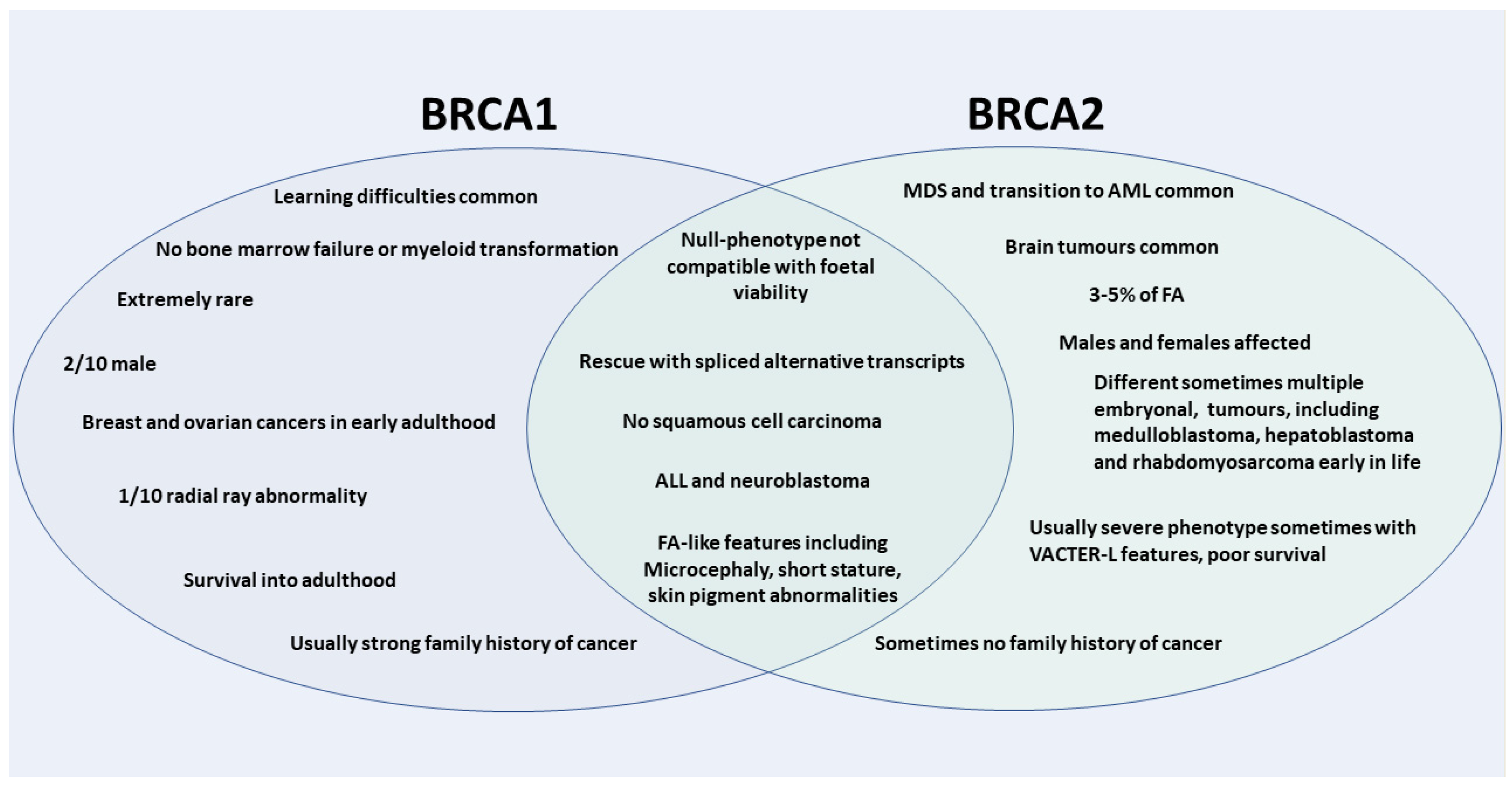
2. FA Caused by FANCD1/BRCA2 Pathogenic Variants: Severe Phenotype with Early Embryonal Malignancies
3. FA Caused by BRCA1/FANCS Pathogenic Variants: Distinct Clinical Phenotype and Cancer Spectrum
4. BRCA1 and BRCA2 PGVs in Non-FA Childhood Cancer
5. BRCA1/2 PGVs in FA-Associated, and Non-Syndromic Cancer: Implications for Management
6. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schneider, M.; Chandler, K.; Tischkowitz, M.; Meyer, S. Fanconi anaemia: Genetics, molecular biology, and cancer-implications for clinical management in children and adults. Clin. Genet. 2015, 88, 13–24. [Google Scholar] [CrossRef]
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome instability syndromes. Nat. Rev. Dis. Primers 2019, 5, 64. [Google Scholar] [CrossRef]
- Huck, K.; Hanenberg, H.; Gudowius, S.; Fenk, R.; Kalb, R.; Neveling, K.; Betz, B.; Niederacher, D.; Haas, R.; Gobel, U.; et al. Delayed diagnosis and complications of Fanconi anaemia at advanced age—A paradigm. Br. J. Haematol. 2006, 133, 188–197. [Google Scholar] [CrossRef]
- Neveling, K.; Endt, D.; Hoehn, H.; Schindler, D. Genotype-phenotype correlations in Fanconi anemia. Mutat. Res. 2009, 668, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef] [PubMed]
- Fanconi, G. Familiare infantile perniziosaartige anamie (pernizioses blutbild und konstitution). Jahrb. Kinderheilk 1927, 117, 257–280. [Google Scholar]
- Knies, K.; Inano, S.; Ramirez, M.J.; Ishiai, M.; Surralles, J.; Takata, M.; Schindler, D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Investig. 2017, 127, 3013–3027. [Google Scholar] [CrossRef] [PubMed]
- Niraj, J.; Farkkila, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Wang, A.T.; Kim, T.; Wagner, J.E.; Conti, B.A.; Lach, F.P.; Huang, A.L.; Molina, H.; Sanborn, E.M.; Zierhut, H.; Cornes, B.K.; et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol. Cell 2015, 59, 478–490. [Google Scholar] [CrossRef]
- Langevin, F.; Crossan, G.P.; Rosado, I.V.; Arends, M.J.; Patel, K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011, 475, 53–58. [Google Scholar] [CrossRef]
- Rosado, I.V.; Langevin, F.; Crossan, G.P.; Takata, M.; Patel, K.J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 2011, 18, 1432–1434. [Google Scholar] [CrossRef]
- Shakeel, S.; Rajendra, E.; Alcon, P.; O’Reilly, F.; Chorev, D.S.; Maslen, S.; Degliesposti, G.; Russo, C.J.; He, S.; Hill, C.H.; et al. Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 2019, 575, 234–237. [Google Scholar] [CrossRef]
- Chen, C.C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Tian, L.; Kahkonen, M.; Schwartzentruber, J.; Kircher, M.; Majewski, J.; Dyment, D.A.; Innes, A.M. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Rickman, K.A.; Noonan, R.J.; Lach, F.P.; Sridhar, S.; Wang, A.T.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846. [Google Scholar] [CrossRef]
- Foulkes, W.D. Inherited susceptibility to common cancers. N. Engl. J. Med. 2008, 359, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Targeted mutations of breast cancer susceptibility gene homologs in mice: Lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 1997, 11, 1226–1241. [Google Scholar] [CrossRef] [PubMed]
- Evers, B.; Jonkers, J. Mouse models of BRCA1 and BRCA2 deficiency: Past lessons, current understanding and future prospects. Oncogene 2006, 25, 5885–5897. [Google Scholar] [CrossRef]
- Meyer, S.; Tischkowitz, M.; Chandler, K.; Gillespie, A.; Birch, J.M.; Evans, D.G. Fanconi anaemia, BRCA2 mutations and childhood cancer: A developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J. Med. Genet. 2014, 51, 71–75. [Google Scholar] [CrossRef]
- Feben, C.; Spencer, C.; Lochan, A.; Laing, N.; Fieggen, K.; Honey, E.; Wainstein, T.; Krause, A. Biallelic BRCA2 mutations in two black South African children with Fanconi anaemia. Fam. Cancer 2017, 16, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Offit, K.; Levran, O.; Mullaney, B.; Mah, K.; Nafa, K.; Batish, S.D.; Diotti, R.; Schneider, H.; Deffenbaugh, A.; Scholl, T.; et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J. Natl. Cancer Inst. 2003, 95, 1548–1551. [Google Scholar] [CrossRef]
- Radulovic, I.; Kuechler, A.; Schundeln, M.M.; Paulussen, M.; von Neuhoff, N.; Reinhardt, D.; Hanenberg, H. A homozygous nonsense mutation early in exon 5 of BRCA2 is associated with very severe Fanconi anemia. Eur. J. Med. Genet. 2021, 64, 104260. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.; Renwick, A.; Seal, S.; Baskcomb, L.; Barfoot, R.; Jayatilake, H.; Pritchard-Jones, K.; Stratton, M.R.; Ridolfi-Luthy, A.; Rahman, N.; et al. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J. Med. Genet. 2005, 42, 147–151. [Google Scholar] [CrossRef][Green Version]
- Malric, A.; Defachelles, A.S.; Leblanc, T.; Lescoeur, B.; Lacour, B.; Peuchmaur, M.; Maurage, C.A.; Pierron, G.; Guillemot, D.; d’Enghien, C.D.; et al. Fanconi anemia and solid malignancies in childhood: A national retrospective study. Pediatr. Blood Cancer 2015, 62, 463–470. [Google Scholar] [CrossRef]
- Dodgshun, A.J.; Sexton-Oates, A.; Saffery, R.; Sullivan, M.J. Biallelic FANCD1/BRCA2 mutations predisposing to glioblastoma multiforme with multiple oncogenic amplifications. Cancer Genet. 2016, 209, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Degrolard-Courcet, E.; Sokolowska, J.; Padeano, M.M.; Guiu, S.; Bronner, M.; Chery, C.; Coron, F.; Lepage, C.; Chapusot, C.; Loustalot, C.; et al. Development of primary early-onset colorectal cancers due to biallelic mutations of the FANCD1/BRCA2 gene. Eur. J. Hum. Genet. 2014, 22, 979–987. [Google Scholar] [CrossRef]
- Alter, B.P.; Rosenberg, P.S. VACTERL-H Association and Fanconi Anemia. Mol. Syndromol. 2013, 4, 87–93. [Google Scholar] [CrossRef]
- Alter, B.P.; Rosenberg, P.S.; Brody, L.C. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J. Med. Genet. 2007, 44, 1–9. [Google Scholar] [CrossRef]
- Kopic, S.; Eirich, K.; Schuster, B.; Hanenberg, H.; Varon-Mateeva, R.; Rittinger, O.; Schimpl, G.; Schindler, D.; Jones, N. Hepatoblastoma in a 4-year-old girl with Fanconi anaemia. Acta Paediatr. 2011, 100, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Fergusson, W.D.; Oostra, A.B.; Medhurst, A.L.; Waisfisz, Q.; de Winter, J.P.; Chen, F.; Carr, T.F.; Clayton-Smith, J.; Clancy, T.; et al. A cross-linker-sensitive myeloid leukemia cell line from a 2-year-old boy with severe Fanconi anemia and biallelic FANCD1/BRCA2 mutations. Genes Chromosomes Cancer 2005, 42, 404–415. [Google Scholar] [CrossRef]
- Dewire, M.D.; Ellison, D.W.; Patay, Z.; McKinnon, P.J.; Sanders, R.P.; Gajjar, A. Fanconi anemia and biallelic BRCA2 mutation diagnosed in a young child with an embryonal CNS tumor. Pediatr. Blood Cancer 2009, 53, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Caburet, S.; Heddar, A.; Dardillac, E.; Creux, H.; Lambert, M.; Messiaen, S.; Tourpin, S.; Livera, G.; Lopez, B.S.; Misrahi, M. Homozygous hypomorphic BRCA2 variant in primary ovarian insufficiency without cancer or Fanconi anaemia trait. J. Med. Genet. 2020, 58, 125–134. [Google Scholar] [CrossRef]
- Oddoux, C.; Struewing, J.P.; Clayton, C.M.; Neuhausen, S.; Brody, L.C.; Kaback, M.; Haas, B.; Norton, L.; Borgen, P.; Jhanwar, S.; et al. The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1%. Nat. Genet. 1996, 14, 188–190. [Google Scholar] [CrossRef]
- Thorlacius, S.; Olafsdottir, G.; Tryggvadottir, L.; Neuhausen, S.; Jonasson, J.G.; Tavtigian, S.V.; Tulinius, H.; Ogmundsdottir, H.M.; Eyfjord, J.E. A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nat. Genet. 1996, 13, 117–119. [Google Scholar] [CrossRef]
- Biswas, K.; Das, R.; Alter, B.P.; Kuznetsov, S.G.; Stauffer, S.; North, S.L.; Burkett, S.; Brody, L.C.; Meyer, S.; Byrd, R.A.; et al. A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell-based assay. Blood 2011, 118, 2430–2442. [Google Scholar] [CrossRef]
- Thirthagiri, E.; Klarmann, K.D.; Shukla, A.K.; Southon, E.; Biswas, K.; Martin, B.K.; North, S.L.; Magidson, V.; Burkett, S.; Haines, D.C.; et al. BRCA2 minor transcript lacking exons 4–7 supports viability in mice and may account for survival of humans with a pathogenic biallelic mutation. Hum. Mol. Genet. 2016, 25, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Lipton, G.B.; Stauffer, S.; Sullivan, T.; Cleveland, L.; Southon, E.; Reid, S.; Magidson, V.; Iversen, E.S., Jr.; Sharan, S.K. A computational model for classification of BRCA2 variants using mouse embryonic stem cell-based functional assays. NPJ Genom. Med. 2020, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Stevens, A.; Paredes, R.; Schneider, M.; Walker, M.J.; Williamson, A.J.K.; Gonzalez-Sanchez, M.B.; Smetsers, S.; Dalal, V.; Teng, H.Y.; et al. Acquired cross-linker resistance associated with a novel spliced BRCA2 protein variant for molecular phenotyping of BRCA2 disruption. Cell Death Dis. 2017, 8, e2875. [Google Scholar] [CrossRef]
- Chirita-Emandi, A.; Andreescu, N.; Popa, C.; Mihailescu, A.; Riza, A.L.; Plesea, R.; Ioana, M.; Arghirescu, S.; Puiu, M. Biallelic variants in BRCA1 gene cause a recognisable phenotype within chromosomal instability syndromes reframed as BRCA1 deficiency. J. Med. Genet. 2020, 58, 648–652. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 2014, 343, 1470–1475. [Google Scholar] [CrossRef]
- Tacconi, E.M.; Lai, X.; Folio, C.; Porru, M.; Zonderland, G.; Badie, S.; Michl, J.; Sechi, I.; Rogier, M.; Matia Garcia, V.; et al. BRCA1 and BRCA2 tumor suppressors protect against endogenous acetaldehyde toxicity. EMBO Mol. Med. 2017, 9, 1398–1414. [Google Scholar] [CrossRef]
- Ducy, M.; Sesma-Sanz, L.; Guitton-Sert, L.; Lashgari, A.; Gao, Y.; Brahiti, N.; Rodrigue, A.; Margaillan, G.; Caron, M.C.; Cote, J.; et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 2019, 44, 226–240. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.; Steinberg-Shemer, O.; Unal, S.; Casadei, S.; Walsh, T.; Gumruk, F.; Shalev, S.; Shimamura, A.; Akarsu, N.A.; Tamary, H.; et al. Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene BRCA1. Proc. Natl. Acad. Sci. USA 2018, 115, 5241–5246. [Google Scholar] [CrossRef]
- Hakem, R.; de la Pompa, J.L.; Sirard, C.; Mo, R.; Woo, M.; Hakem, A.; Wakeham, A.; Potter, J.; Reitmair, A.; Billia, F.; et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 1996, 85, 1009–1023. [Google Scholar] [CrossRef]
- Gowen, L.C.; Johnson, B.L.; Latour, A.M.; Sulik, K.K.; Koller, B.H. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat. Genet. 1996, 12, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Tang, J.; Stopfer, J.; Lilli, D.R.; Hamel, N.; Tischkowitz, M.; Monteiro, A.N.; Messick, T.E.; Powers, J.; Yonker, A.; et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013, 3, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Freire, B.L.; Homma, T.K.; Funari, M.F.A.; Lerario, A.M.; Leal, A.M.; Velloso, E.; Malaquias, A.C.; Jorge, A.A.L. Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients. Eur. J. Med. Genet. 2018, 61, 130–133. [Google Scholar] [CrossRef]
- Kwong, A.; Ho, C.Y.S.; Shin, V.Y.; Au, C.H.; Chan, T.L.; Ma, E.S.K. A Case Report of Germline Compound Heterozygous Mutations in the BRCA1 Gene of an Ovarian and Breast Cancer Patient. Int. J. Mol. Sci. 2021, 22, 889. [Google Scholar] [CrossRef] [PubMed]
- Keupp, K.; Hampp, S.; Hubbel, A.; Maringa, M.; Kostezka, S.; Rhiem, K.; Waha, A.; Wappenschmidt, B.; Pujol, R.; Surralles, J.; et al. Biallelic germline BRCA1 mutations in a patient with early onset breast cancer, mild Fanconi anemia-like phenotype, and no chromosome fragility. Mol. Genet. Genom. Med. 2019, 7, e863. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A.; Stopfer, J.E.; Erlichman, J.; Davidson, R.; Nathanson, K.L.; Domchek, S.M. Childhood cancer in families with and without BRCA1 or BRCA2 mutations ascertained at a high-risk breast cancer clinic. Cancer Biol. Ther. 2006, 5, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, S.; Borg, A.; Kristoffersson, U.; Nilbert, M.; Wiebe, T.; Olsson, H. Higher occurrence of childhood cancer in families with germline mutations in BRCA2, MMR and CDKN2A genes. Fam. Cancer 2008, 7, 331–337. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Sinilnikova, O.M.; Simard, J.; Leone, M.; Dumont, M.; Neuhausen, S.L.; Struewing, J.P.; Stoppa-Lyonnet, D.; Barjhoux, L.; Hughes, D.J.; et al. RAD51 135G-->C modifies breast cancer risk among BRCA2 mutation carriers: Results from a combined analysis of 19 studies. Am. J. Hum. Genet. 2007, 81, 1186–1200. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef]
- Qin, N.; Wang, Z.; Liu, Q.; Song, N.; Wilson, C.L.; Ehrhardt, M.J.; Shelton, K.; Easton, J.; Mulder, H.; Kennetz, D.; et al. Pathogenic Germline Mutations in DNA Repair Genes in Combination with Cancer Treatment Exposures and Risk of Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer. J. Clin. Oncol. 2020, 38, 2728–2740. [Google Scholar] [CrossRef]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients with Osteosarcoma. JAMA Oncol. 2020, 6, 724–734. [Google Scholar] [CrossRef]
- Muskens, I.S.; de Smith, A.J.; Zhang, C.; Hansen, H.M.; Morimoto, L.; Metayer, C.; Ma, X.; Walsh, K.M.; Wiemels, J.L. Germline cancer predisposition variants and pediatric glioma: A population-based study in California. Neuro. Oncol. 2020, 22, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Light, N.; Subasri, V.; Young, E.L.; Wegman-Ostrosky, T.; Barkauskas, D.A.; Hall, D.; Lupo, P.J.; Patidar, R.; Maese, L.D.; et al. Pathogenic Germline Variants in Cancer Susceptibility Genes in Children and Young Adults with Rhabdomyosarcoma. JCO Precis. Oncol. 2021, 5, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Grzymski, J.J.; Elhanan, G.; Morales Rosado, J.A.; Smith, E.; Schlauch, K.A.; Read, R.; Rowan, C.; Slotnick, N.; Dabe, S.; Metcalf, W.J.; et al. Population genetic screening efficiently identifies carriers of autosomal dominant diseases. Nat. Med. 2020, 26, 1235–1239. [Google Scholar] [CrossRef]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugieres, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Li, H.; Sisoudiya, S.D.; Martin-Giacalone, B.A.; Khayat, M.M.; Dugan-Perez, S.; Marquez-Do, D.A.; Scheurer, M.E.; Muzny, D.; Boerwinkle, E.; Gibbs, R.A.; et al. Germline Cancer Predisposition Variants in Pediatric Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Natl. Cancer Inst. 2021, 113, 875–883. [Google Scholar] [CrossRef]
- Wang, Z.; Wilson, C.L.; Armstrong, G.T.; Hudson, M.M.; Zhang, J.; Nichols, K.E.; Robison, L.L. Association of Germline BRCA2 Mutations with the Risk of Pediatric or Adolescent Non-Hodgkin Lymphoma. JAMA Oncol. 2019, 5, 1362–1364. [Google Scholar] [CrossRef] [PubMed]
- Haude, K.; McCarthy Veach, P.; LeRoy, B.; Zierhut, H. Factors Influencing the Decision-Making Process and Long-Term Interpersonal Outcomes for Parents Who Undergo Preimplantation Genetic Diagnosis for Fanconi Anemia: A Qualitative Investigation. J. Genet. Couns. 2017, 26, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Zierhut, H.A.; Tryon, R.; Sanborn, E.M. Genetic counseling for Fanconi anemia: Crosslinking disciplines. J. Genet. Couns. 2014, 23, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Tesch, B.A.; Gawande, R.S.; Zhang, L.; MacMillan, M.L.; Nascene, D.R. Fanconi anemia: Correlating central nervous system malformations and genetic complementation groups. Pediatr. Radiol. 2017, 47, 868–876. [Google Scholar] [CrossRef]
- Stivaros, S.M.; Alston, R.; Wright, N.B.; Chandler, K.; Bonney, D.; Wynn, R.F.; Will, A.M.; Punekar, M.; Loughran, S.; Kilday, J.P.; et al. Central nervous system abnormalities in Fanconi anaemia: Patterns and frequency on magnetic resonance imaging. Br. J. Radiol. 2015, 88, 20150088. [Google Scholar] [CrossRef]
- Mitchell, R.; Wagner, J.E.; Hirsch, B.; De For, T.E.; Zierhut, H.; MacMillan, M.L. Haematopoietic cell transplantation for acute leukaemia and advanced myelodysplastic syndrome in Fanconi anaemia. Br. J. Haematol. 2014, 164, 384–395. [Google Scholar] [CrossRef]
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmana, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef] [PubMed]
- Brenner, J.C.; Feng, F.Y.; Han, S.; Patel, S.; Goyal, S.V.; Bou-Maroun, L.M.; Liu, M.; Lonigro, R.; Prensner, J.R.; Tomlins, S.A.; et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012, 72, 1608–1613. [Google Scholar] [CrossRef]
- Fackenthal, J.D.; Yoshimatsu, T.; Zhang, B.; de Garibay, G.R.; Colombo, M.; De Vecchi, G.; Ayoub, S.C.; Lal, K.; Olopade, O.I.; Vega, A.; et al. Naturally occurring BRCA2 alternative mRNA splicing events in clinically relevant samples. J. Med. Genet. 2016, 53, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Gaildrat, P.; Krieger, S.; Di Giacomo, D.; Abdat, J.; Revillion, F.; Caputo, S.; Vaur, D.; Jamard, E.; Bohers, E.; Ledemeney, D.; et al. Multiple sequence variants of BRCA2 exon 7 alter splicing regulation. J. Med. Genet. 2012, 49, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Cassidy, L.D.; Pisupati, V.; Jonasson, J.G.; Bjarnason, H.; Eyfjord, J.E.; Karreth, F.A.; Lim, M.; Barber, L.M.; Clatworthy, S.A.; et al. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 2010, 18, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.F.; Kennedy, J.; Harlan, M.; Kentsis, A.; Shukla, N.; Musinsky, J.; Roberts, S.; Kung, A.L.; Robson, M.; Kushner, B.H.; et al. Germline BRCA2 mutations detected in pediatric sequencing studies impact parents’ evaluation and care. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001925. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| BRCA1 Variants | BRCA2 Variants | Ref | |
|---|---|---|---|
| Long Term Survivor Study | 14/4402 | 20/4402 | Qin et al. 2020 [59] |
| Osteosarcoma | 3/1440 | 8/1440 | Mirabello et al. 2020 [60] |
| Paediatric Glioma | 1/220 | 1/220 | Muskens et al. 2020 [61] |
| Rhabdomyosarcoma | 1/615 | 6/615 | Li et al. 2021 [65] |
| Paed and adol. non-Hodgkins Lymphoma | not included | 13/1380 | Wang et al. 2019 [67] |
| Medulloblastoma | - | 11/1022 | Waszak et al. 2018 [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodward, E.R.; Meyer, S. Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes 2021, 12, 1520. https://doi.org/10.3390/genes12101520
Woodward ER, Meyer S. Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes. 2021; 12(10):1520. https://doi.org/10.3390/genes12101520
Chicago/Turabian StyleWoodward, Emma R., and Stefan Meyer. 2021. "Fanconi Anaemia, Childhood Cancer and the BRCA Genes" Genes 12, no. 10: 1520. https://doi.org/10.3390/genes12101520
APA StyleWoodward, E. R., & Meyer, S. (2021). Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes, 12(10), 1520. https://doi.org/10.3390/genes12101520