
Fine Mapping of the Mouse Ath28 Locus Yields Three Atherosclerosis Modifying Sub-Regions


Abstract
1. Introduction
2. Materials and Methods
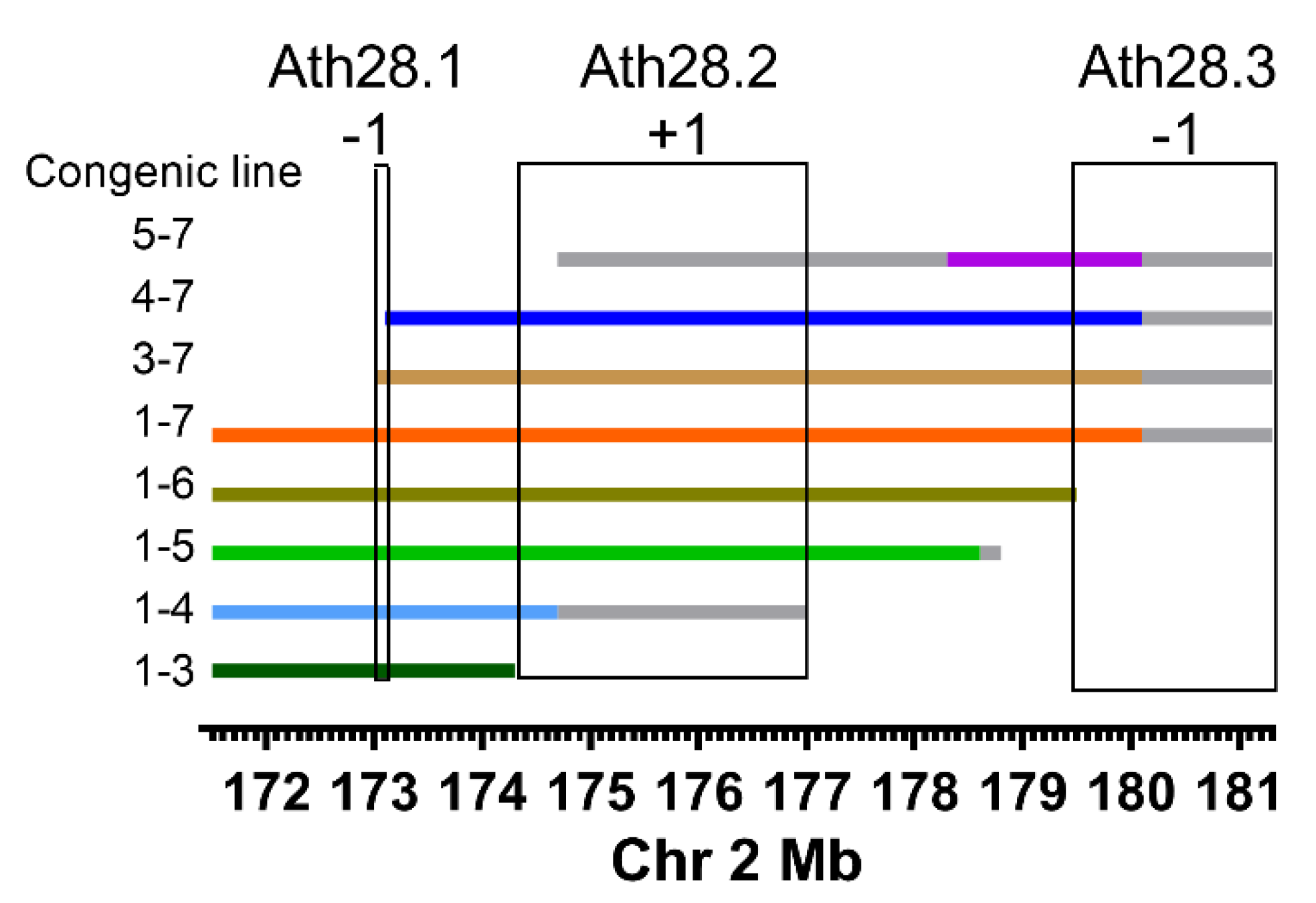
2.1. Ath28 Congenic Lines
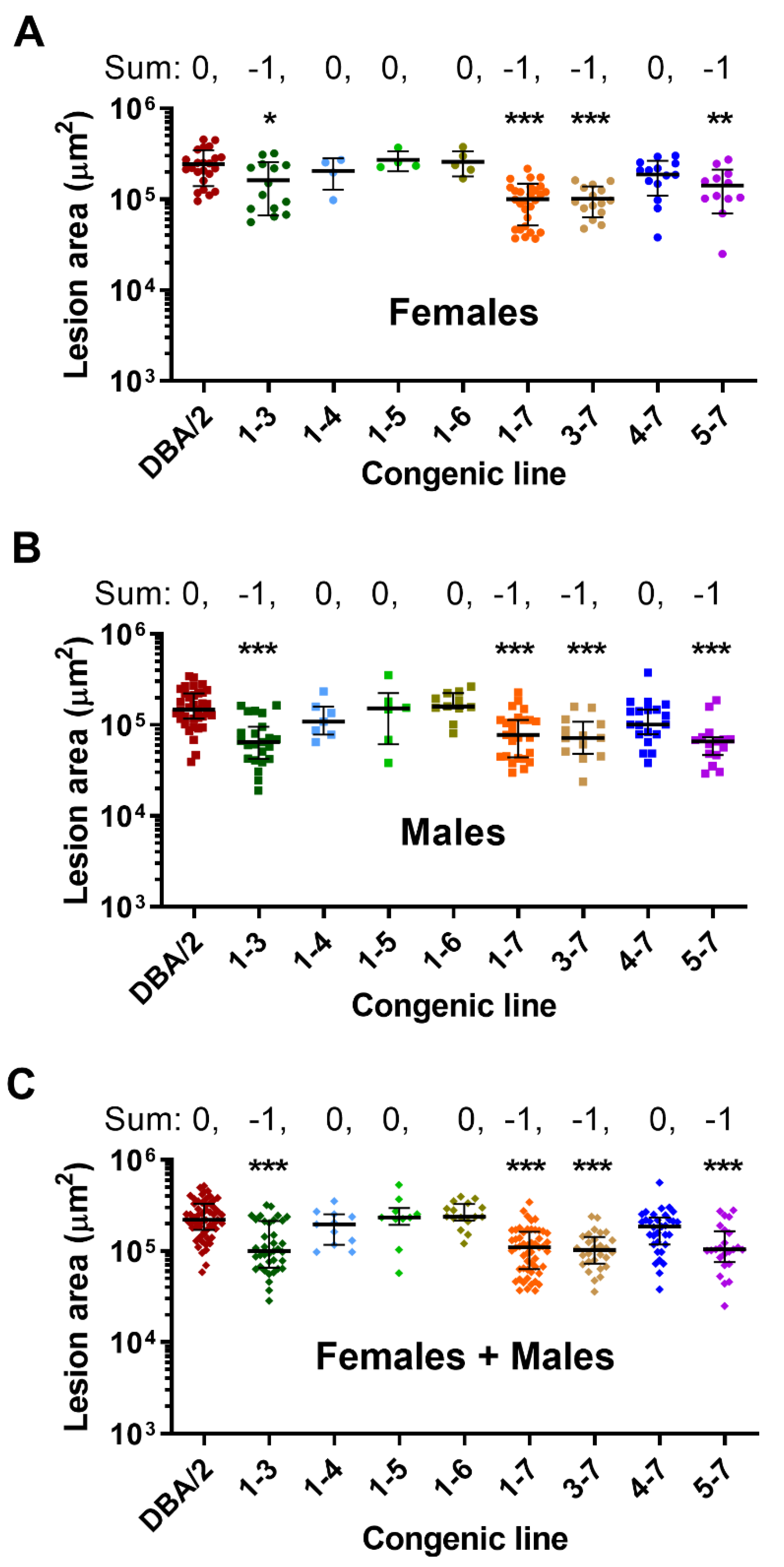
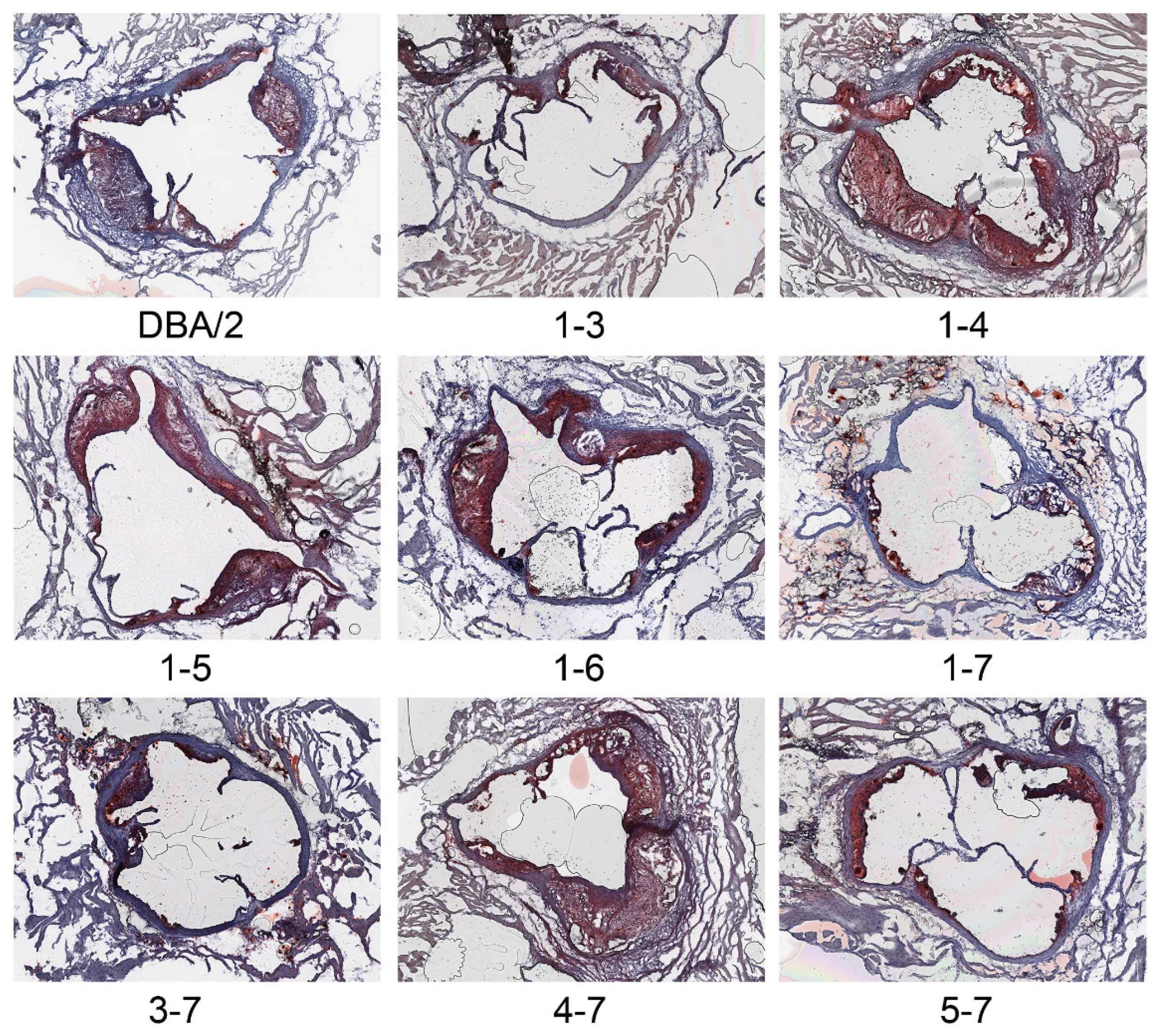
2.2. Phenotypic Analyses
2.3. Statistical Analysis
2.4. Bioinformatic Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Getz, G.S.; Reardon, C.A. Use of mouse models in atherosclerosis research. In Methods in Molecular Biology; Andres, V., Dorado, B., Eds.; Springer: New York, NY, USA, 2015; pp. 1–16. [Google Scholar]
- Smith, J.D. Quantitative trait locus mapping to identify genes for complex traits in Mice. In Molecular Biomethods Handbook; Walker, J., Rapley, R., Eds.; Springer: Totowa, NJ, USA, 2008; pp. 257–268. ISBN 9781603273701. [Google Scholar]
- Rodríguez, J.M.; Wolfrum, S.; Robblee, M.; Chen, K.Y.; Gilbert, Z.N.; Choi, J.-H.; Teupser, D.; Breslow, J.L. Altered expression of raet1e, a major histocompatibility complex class 1-like molecule, underlies the atherosclerosis modifier locus ath11 10b. Circ. Res. 2013, 113, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Kayashima, Y.; Makhanova, N.; Maeda, N. DBA/2J Haplotype on Distal Chromosome 2 Reduces Mertk Expression, Restricts Efferocytosis, and Increases Susceptibility to Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e82–e91. [Google Scholar] [CrossRef][Green Version]
- Smith, J.D.; James, D.; Dansky, H.M.; Wittkowski, K.M.; Moore, K.J.; Breslow, J.L. In silico quantitative trait locus map for atherosclerosis susceptibility in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Smith, J.D. Genetic-genomic replication to identify candidate mouse atherosclerosis modifier genes. J. Am. Heart Assoc. 2013, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Baglione, J.; Smith, J.D. Quantitative Assay for Mouse Atherosclerosis in the Aortic Root. Methods Mol. Med. 2006, 129, 83–96. [Google Scholar] [CrossRef]
- Han, J.; Robinet, P.; Ritchey, B.; Andro, H.; Smith, J.D. Confirmation of Ath26 locus on chromosome 17 and identification of Cyp4f13 as an atherosclerosis modifying gene. Atherosclerosis 2019, 286, 71–78. [Google Scholar] [CrossRef]
- Smith, J.D.; Bhasin, J.M.; Baglione, J.; Settle, M.; Xu, Y.; Barnard, J. Atherosclerosis susceptibility loci identified from a strain intercross of apolipoprotein E-deficient mice via a high-density genome scan. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 507–603. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Yazbek, S.N.; Buchner, D.A.; Geisinger, J.M.; Burrage, L.C.; Spiezio, S.H.; Zentner, G.E.; Hsieh, C.W.; Scacheri, P.C.; Croniger, C.M.; Nadeau, J.H. Deep congenic analysis identifies many strong, context-dependent QTLs, one of which, Slc35b4, regulates obesity and glucose homeostasis. Genome Res. 2011, 21, 1065–1073. [Google Scholar] [CrossRef]
- Sohrabi, Y.; Reinecke, H. RIPK1 targeting protects against obesity and atherosclerosis. Trends Endocrinol. Metab. 2021, 32, 420–422. [Google Scholar] [CrossRef]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef]
- Karunakaran, D.; Nguyen, M.-A.; Geoffrion, M.; Vreeken, D.; Lister, Z.; Cheng, H.S.; Otte, N.; Essebier, P.; Wyatt, H.; Kandiah, J.W.; et al. RIPK1 Expression Associates with Inflammation in Early Atherosclerosis in Humans and Can be Therapeutically Silenced to Reduce NF-κB Activation and Atherogenesis in Mice. Circulation 2020, 143, 163–177. [Google Scholar] [CrossRef]
- Zhao, T.X.; Mallat, Z. Targeting the Immune System in Atherosclerosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 1691–1706. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Seenappa, V.; Joshi, M.; Satyamoorthy, K. Intricate Regulation of Phosphoenolpyruvate Carboxykinase (PEPCK) Isoforms in Normal Physiology and DiseaseNo Title. Curr. Mol. Med. 2019, 19, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, P.; Garcia-Rios, A.; Delgado-Lista, J.; Gjelstad, I.M.F.; Gibney, J.; Kieć-Wilk, B.; Camargo, A.; Helal, O.; Karlström, B.; Blaak, E.E.; et al. Gene-nutrient interactions on the phosphoenolpyruvate carboxykinase influence insulin sensitivity in metabolic syndrome subjects. Clin. Nutr. 2013, 32, 630–635. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, T.; Xie, P.; Wang, L.; Chen, H.; Cheng, Z.; Zhou, J. PMEPA1 interference activates PTEN/PI3K/AKT, thereby inhibiting the proliferation, invasion and migration of pancreatic cancer cells and enhancing the sensitivity to gemcitabine and cisplatin. Drug Dev. Res. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Xu, X.; Nishioka, K.; Matsuhisa, F.; Kitajima, S.; Kukita, T.; Murayama, M.; Urano, Y.; Miyamoto, H.; Mawatari, M.; et al. PMEPA1 and NEDD4 control the proton production of osteoclasts by regulating vesicular trafficking. FASEB J. 2021, 35, e21281. [Google Scholar] [CrossRef] [PubMed]
- von Scheidt, M.; Zhao, Y.; Kurt, Z.; Pan, C.; Zeng, L.; Yang, X.; Schunkert, H.; Lusis, A.J. Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metab. 2017, 25, 248–261. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mouse Line | Body Wt g Mean ± SD (n) | p-Value a | Total Cholesterol mg/dL Median [IQR] (n) | p-Value b | HDL Cholesterol mg/dL Median [IQR] (n) | p-Value b |
|---|---|---|---|---|---|---|
| Males | ||||||
| DBA/2 | 26.2 ± 2.5 (39) | 1099 (888–1206) (36) | 27 (16–50.2) (27) | |||
| 1-7 | 26.3 ± 2.9 (28) | ns | 1089 (951–1217) (28) | ns | 43.2 (30.2–62) (22) | ns |
| 1-6 | 27.0 ± 1.5 (11) | ns | 1173 (1016–1345) (11) | ns | 54.7 (38–89.1) (8) | * |
| 1-5 | 24.2 ± 3.5 (7) | ns | 1056 (883–1164) (7) | ns | 43.6 (36.5–68) (7) | ns |
| 1-4 | 26.9 ± 2.0 (15) | ns | 1077 (870–1120) (15) | ns | 38.7 (29.5–50.3) (14) | ns |
| 1-3 | 26.6 ± 2.1 (24) | ns | 1031 (891–1151) (24) | ns | 51 (38.4–60.4) (24) | * |
| 3-7 | 27.2 ± 2.0 (12) | ns | 1077 (1000–1131) (12) | ns | 30.5 (26.1–43.1) (10) | ns |
| 4-7 | 24.1 ± 2.2 (22) | ** | 1111 (1003–1244) (18) | ns | 37.2 (26.6–56.6) (16) | ns |
| 5-7 | 24.7 ± 1.7 (14) | ns | 1016 (934–1042) (15) | ns | 44.8 (26.6–63.3) (14) | ns |
| Females | ||||||
| DBA/2 | 21.2 ± 2.2 (25) | 1123 (935–1245) (25) | 29.3 (12.6–39.6) (23) | |||
| 1-7 | 22.2 ±1.8 (28) | ns | 1222 (962–1447) (28) | ns | 40.2 (24.2–63.1) (21) | ns |
| 1-6 | 23.8 ±2.3 (5) | ns | 1242 (1187–1270) (5) | ns | 73.3 (55.7–75.3) (5) | * |
| 1-5 | 22.5 ±1.9 (7) | ns | 1151 (813–1170) (7) | ns | 47.5 (39.7–65.8) (7) | ns |
| 1-4 | 20.5 ±2.0 (8) | ns | 890 (797–933) (8) | * | 29.2 (18.8–44.6) (8) | ns |
| 1-3 | 22.4 ±3.0 (16) | ns | 951 (839–1086) (16) | ns | 21.7 (14.7–57.7) (15) | ns |
| 3-7 | 23.3 ±1.8 (15) | ns | 1230 (1064–1501) (15) | ns | 23.5 (17.1–48.3) (15) | ns |
| 4-7 | 21.4 ±1.9 (19) | ns | 1147 (1061–1235) (17) | ns | 37.2 (26.6–56.6) (16) | ns |
| 5-7 | 21.2 ±1.7 (12) | ns | 956 (893–1116) (12) | ns | 33.2 (23–52.8) (12) | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Ritchey, B.; Opoku, E.; Smith, J.D. Fine Mapping of the Mouse Ath28 Locus Yields Three Atherosclerosis Modifying Sub-Regions. Genes 2022, 13, 70. https://doi.org/10.3390/genes13010070
Han J, Ritchey B, Opoku E, Smith JD. Fine Mapping of the Mouse Ath28 Locus Yields Three Atherosclerosis Modifying Sub-Regions. Genes. 2022; 13(1):70. https://doi.org/10.3390/genes13010070
Chicago/Turabian StyleHan, Juying, Brian Ritchey, Emmanuel Opoku, and Jonathan D. Smith. 2022. "Fine Mapping of the Mouse Ath28 Locus Yields Three Atherosclerosis Modifying Sub-Regions" Genes 13, no. 1: 70. https://doi.org/10.3390/genes13010070
APA StyleHan, J., Ritchey, B., Opoku, E., & Smith, J. D. (2022). Fine Mapping of the Mouse Ath28 Locus Yields Three Atherosclerosis Modifying Sub-Regions. Genes, 13(1), 70. https://doi.org/10.3390/genes13010070