
Influence of Haptoglobin Polymorphism on Stroke in Sickle Cell Disease Patients

 ,
, 
 , ,
, , 
Abstract
:1. Introduction
1.1. Sickle Cell Disease
1.2. Haptoglobin Polymorphism
1.3. Critical Role of Haptoglobin
1.4. Blood Exchange Transfusions
2. Methods
3. Current Evidence Regarding Hp Genotypes and Stroke
4. Discussion
Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piel, F.B.; Patil, A.P.; Howes, R.E.; Nyangiri, O.A.; Gething, P.W.; Dewi, M.; Temperley, W.H.; Williams, T.N.; Weatherall, D.J.; Hay, S.I. Global epidemiology of sickle haemoglobin in neonates: A contemporary geostatistical model-based map and population estimates. Lancet 2013, 381, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, P.; Sarmah, D.; Dave, K.R.; Goswami, A.; Watanabe, M.; Wang, X.; Kalia, K.; Plesnila, N.; Yavagal, D.R.; Alvarez, O. Stroke and stroke prevention in sickle cell anemia in developed and selected developing countries. J. Neurol. Sci. 2021, 427, 117510. [Google Scholar] [CrossRef]
- Sedrak, A.; Kondamudi, N.P. Sickle Cell Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Ashouri, R.; Fangman, M.; Burris, A.; Ezenwa, M.O.; Wilkie, D.J.; Doré, S. Critical role of hemopexin mediated cytoprotection in the pathophysiology of sickle cell disease. Int. J. Mol. Sci. 2021, 22, 6408. [Google Scholar] [CrossRef]
- Switzer, J.A.; Hess, D.C.; Nichols, F.T.; Adams, R.J. Pathophysiology and treatment of stroke in sickle-cell disease: Present and future. Lancet Neurol. 2006, 5, 501–512. [Google Scholar] [CrossRef]
- Carter, K.; Worwood, M. Haptoglobin: A review of the major allele frequencies worldwide and their association with diseases. Int. J. Lab. Hematol. 2007, 29, 92–110. [Google Scholar] [CrossRef]
- Wobeto, V.P.D.A.; Zaccariotto, T.R.; Sonati, M.D.F. Polymorphism of human haptoglobin and its clinical importance. Genet. Mol. Biol. 2008, 31, 602–620. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, L.C.; Miranda-Vilela, A.L.; Hiragi, C.D.O.; Ribeiro, I.F.; Daldegan, M.B.; Grisolia, C.K.; dos Santos-Neto, L.L. Haptoglobin and myeloperoxidase (-G463A) gene polymorphisms in Brazilian sickle cell patients with and without secondary iron overload. Blood Cells Mol. Dis. 2014, 52, 95–107. [Google Scholar] [CrossRef]
- Atkinson, S.H.; Rockett, K.; Sirugo, G.; Bejon, P.A.; Fulford, A.; O’Connell, M.A.; Bailey, R.; Kwiatkowski, D.P.; Prentice, A.M. Seasonal childhood anaemia in West Africa is associated with the haptoglobin 2-2 genotype. PLoS Med. 2006, 3, e172. [Google Scholar] [CrossRef] [Green Version]
- Vardi, M.; Blum, S.; Levy, A.P. Haptoglobin genotype and cardiovascular outcomes in diabetes mellitus-natural history of the disease and the effect of vitamin E treatment. Meta-analysis of the medical literature. Eur. J. Intern. Med. 2012, 23, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, S.L.; Kumar, P.T.; McBride, D.; Zeineddine, H.A.; Leclerc, J.; Choi, H.A.; Dash, P.K.; Grotta, J.; Aronowski, J.; Cardenas, J.C.; et al. Unique contribution of haptoglobin and haptoglobin genotype in aneurysmal subarachnoid hemorrhage. Front. Physiol. 2018, 9, 592. [Google Scholar] [CrossRef]
- Santiago, R.P.; Guarda, C.C.; Figueiredo, C.V.B.; Fiuza, L.M.; Aleluia, M.M.; Adanho, C.S.A.; Carvalho, M.O.S.; Pitanga, T.N.; Zanette, D.L.; Lyra, I.M.; et al. Serum haptoglobin and hemopexin levels are depleted in pediatric sickle cell disease patients. Blood Cells Mol. Dis. 2018, 72, 34–36. [Google Scholar] [CrossRef]
- Yalamanoglu, A.; Deuel, J.W.; Hunt, R.C.; Baek, J.H.; Hassell, K.; Redinius, K.; Irwin, D.C.; Schaer, D.J.; Buehler, P.W. Depletion of haptoglobin and hemopexin promote hemoglobin-mediated lipoprotein oxidation in sickle cell disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L765–L774. [Google Scholar] [CrossRef] [Green Version]
- Saraf, S.L.; Molokie, R.E.; Nouraie, M.; Sable, C.A.; Luchtman-Jones, L.; Ensing, G.J.; Campbell, A.D.; Rana, S.R.; Niu, X.M.; Machado, R.F.; et al. Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatr. Respir. Rev. 2014, 15, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Minniti, C.P.; Sable, C.; Campbell, A.; Rana, S.; Ensing, G.; Dham, N.; Onyekwere, O.; Nouraie, M.; Kato, G.J.; Gladwin, M.T.; et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: Association with hemolysis and hemoglobin oxygen desaturation. Haematologica 2009, 94, 340–347. [Google Scholar] [CrossRef]
- Sachdev, V.; Kato, G.J.; Gibbs, J.S.R.; Barst, R.J.; Machado, R.F.; Nouraie, M.; Hassell, K.L.; Little, J.A.; Schraufnagel, D.E.; Krishnamurti, L.; et al. Walk-PHASST Investigators Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation 2011, 124, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Harari, O.A.; Liao, J.K. NF-κB and innate immunity in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 32–40. [Google Scholar] [CrossRef]
- Schaer, D.J.; Buehler, P.W. Cell-free hemoglobin and its scavenger proteins: New disease models leading the way to targeted therapies. Cold Spring Harb. Perspect. Med. 2013, 3, a013433. [Google Scholar] [CrossRef] [Green Version]
- Akinsheye, I.; Klings, E.S. Sickle cell anemia and vascular dysfunction: The nitric oxide connection. J. Cell. Physiol. 2010, 224, 620–625. [Google Scholar] [CrossRef]
- Quimby, K.R.; Hambleton, I.R.; Landis, R.C. Intravenous infusion of haptoglobin for the prevention of adverse clinical outcome in Sickle Cell Disease. Med. Hypotheses 2015, 85, 424–432. [Google Scholar] [CrossRef]
- Tsitsikas, D.A.; Sirigireddy, B.; Nzouakou, R.; Calvey, A.; Quinn, J.; Collins, J.; Orebayo, F.; Lewis, N.; Todd, S.; Amos, R.J. Safety, tolerability, and outcomes of regular automated red cell exchange transfusion in the management of sickle cell disease. J. Clin. Apher. 2016, 31, 545–550. [Google Scholar] [CrossRef]
- Howard, J. Sickle cell disease: When and how to transfuse. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Andoniadis, M.; Solhpour, A.; Asghar, S.; Fangman, M.; Ashouri, R.; Doré, S. Contribution of various types of transfusion to acute and delayed intracerebral hemorrhage injury. Front. Neurol. 2021, 12, 727569. [Google Scholar] [CrossRef]
- Pierrot-Gallo, B.S.; Vicari, P.; Matsuda, S.S.; Adegoke, S.A.; Mecabo, G.; Figueiredo, M.S. Haptoglobin gene polymorphisms and interleukin-6 and -8 levels in patients with sickle cell anemia. Rev. Bras. Hematol. Hemoter. 2015, 37, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Olatunya, O.S.; Albuquerque, D.M.; Santos, M.N.N.; Kayode, T.S.; Adekile, A.; Costa, F.F. Haptoglobin Gene Polymorphism in Patients with Sickle Cell Anemia: Findings from a Nigerian Cohort Study. Appl. Clin. Genet. 2020, 13, 107–114. [Google Scholar] [CrossRef]
- Willen, S.M.; McNeil, J.B.; Rodeghier, M.; Kerchberger, V.E.; Shaver, C.M.; Bastarache, J.A.; Steinberg, M.H.; DeBaun, M.R.; Ware, L.B. Haptoglobin genotype predicts severe acute vaso-occlusive pain episodes in children with sickle cell anemia. Am. J. Hematol. 2020, 95, E92. [Google Scholar] [CrossRef] [Green Version]
- Adekile, A.D.; Haider, M.Z. Haptoglobin gene polymorphisms in sickle cell disease patients with different βS-globin gene haplotypes. Med. Princ. Pract. 2010, 19, 447–450. [Google Scholar] [CrossRef]
- Cox, S.E.; Makani, J.; Soka, D.; L’Esperence, V.S.; Kija, E.; Dominguez-Salas, P.; Newton, C.R.J.; Birch, A.A.; Prentice, A.M.; Kirkham, F.J. Haptoglobin, α-thalassaemia and glucose-6-phosphate dehydrogenase polymorphisms and risk of abnormal transcranial Doppler among patients with sickle cell anaemia in Tanzania. Br. J. Haematol. 2014, 165, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.N.N.; Bezerra, M.A.C.; Domingues, B.L.T.B.; Zaccariotto, T.R.; Oliveira, D.M.; Costa, F.F.; da Silva Araújo, A.; de Fátima Sonati, M. Haptoglobin genotypes in sickle-cell disease. Genet. Test. Mol. Biomark. 2011, 15, 709–713. [Google Scholar] [CrossRef] [Green Version]
- Hibbert, J.M.; Hsu, L.L.; Bhathena, S.J.; Irune, I.; Sarfo, B.; Creary, M.S.; Gee, B.E.; Mohamed, A.I.; Buchanan, I.D.; Al-Mahmoud, A.; et al. Proinflammatory cytokines and the hypermetabolism of children with sickle cell disease. Exp. Biol. Med. 2005, 230, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Akohoue, S.A.; Shankar, S.; Milne, G.L.; Morrow, J.; Chen, K.Y.; Ajayi, W.U.; Buchowski, M.S. Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr. Res. 2007, 61, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Gonsalves, C.S.; Malik, P.; Kalra, V.K. Placenta growth factor augments endothelin-1 and endothelin-B receptor expression via hypoxia-inducible factor-1 α. Blood 2008, 112, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Lanaro, C.; Franco-Penteado, C.F.; Albuqueque, D.M.; Saad, S.T.O.; Conran, N.; Costa, F.F. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J. Leukoc. Biol. 2009, 85, 235–242. [Google Scholar] [CrossRef]
- Guetta, J.; Strauss, M.; Levy, N.S.; Fahoum, L.; Levy, A.P. Haptoglobin genotype modulates the balance of Th1/Th2 cytokines produced by macrophages exposed to free hemoglobin. Atherosclerosis 2007, 191, 48–53. [Google Scholar] [CrossRef]
- Lindermayr, C.; Durner, J. Interplay of reactive oxygen species and nitric oxide: Nitric oxide coordinates reactive oxygen species homeostasis. Plant Physiol. 2015, 167, 1209–1210. [Google Scholar] [CrossRef] [Green Version]
- Dalan, R.; Liew, H.; Goh, L.L.; Gao, X.; Chew, D.E.; Boehm, B.O.; Leow, M.K.S. The haptoglobin 2-2 genotype is associated with inflammation and carotid artery intima-media thickness. Diab. Vasc. Dis. Res. 2016, 13, 373–376. [Google Scholar] [CrossRef] [Green Version]
- Dahan, I.; Farber, E.; Thauho, N.; Nakhoul, N.; Francis, A.; Awawde, M.; Levy, A.P.; Kim-Shapiro, D.B.; Basu, S.; Nakhoul, F. Interaction between the Haptoglobin 2 Phenotype and Diabetes Mellitus on Systolic Pulmonary Arterial Pressure and Nitric Oxide Bioavailability in Hemodialysis Patients. J. Diabetes Res. 2015, 2015, 613860. [Google Scholar] [CrossRef]
- Sertório, J.T.; Lacchini, R.; Amaral, L.M.; Palei, A.C.T.; Cavalli, R.C.; Sandrim, V.C.; Duarte, G.; Tanus-Santos, J.E. Haptoglobin polymorphism affects nitric oxide bioavailability in preeclampsia. J. Hum. Hypertens. 2013, 27, 349–354. [Google Scholar] [CrossRef]
- Hasson, C.; Veling, L.; Rico, J.; Mhaskar, R. The role of hydroxyurea to prevent silent stroke in sickle cell disease: Systematic review and meta-analysis. Medicine 2019, 98, e18225. [Google Scholar] [CrossRef]
- Kassim, A.A.; Pruthi, S.; Day, M.; Rodeghier, M.; Gindville, M.C.; Brodsky, M.A.; DeBaun, M.R.; Jordan, L.C. Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood 2016, 127, 2038–2040. [Google Scholar] [CrossRef]
- DeBaun, M.R.; Sarnaik, S.A.; Rodeghier, M.J.; Minniti, C.P.; Howard, T.H.; Iyer, R.V.; Inusa, B.; Telfer, P.T.; Kirby-Allen, M.; Quinn, C.T.; et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: Low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood 2012, 119, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Simpore, J.; Pignatelli, S.; Barlati, S.; Musumeci, S. Modification in the frequency of Hb C and Hb S in Burkina Faso: An influence of migratory fluxes and improvement of patient health care. Hemoglobin 2002, 26, 113–120. [Google Scholar] [CrossRef]
- Voskaridou, E.; Ladis, V.; Kattamis, A.; Hassapopoulou, E.; Economou, M.; Kourakli, A.; Maragkos, K.; Kontogianni, K.; Lafioniatis, S.; Vrettou, E.; et al. Greek Haemoglobinopathies Study Group A national registry of haemoglobinopathies in Greece: Deducted demographics, trends in mortality and affected births. Ann. Hematol. 2012, 91, 1451–1458. [Google Scholar] [CrossRef]
- Chandrashekar, V.; Soni, M. Hemoglobin disorders in South India. ISRN Hematol. 2011, 2011, 748939. [Google Scholar] [CrossRef] [Green Version]
- Hulbert, M.L.; McKinstry, R.C.; Lacey, J.L.; Moran, C.J.; Panepinto, J.A.; Thompson, A.A.; Sarnaik, S.A.; Woods, G.M.; Casella, J.F.; Inusa, B.; et al. Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood 2011, 117, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Hamideh, D.; Alvarez, O. Sickle cell disease related mortality in the United States (1999–2009). Pediatr. Blood Cancer 2013, 60, 1482–1486. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The worst things in life are free: The role of free heme in sickle cell disease. Front. Immunol. 2020, 11, 561917. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Hemmaway, C.J.; Telfer, P.; Layton, D.M.; Porter, J.; Awogbade, M.; Mant, T.; Gretler, D.D.; Dufu, K.; Hutchaleelaha, A.; et al. A phase 1/2 ascending dose study and open-label extension study of voxelotor in patients with sickle cell disease. Blood 2019, 133, 1865–1875. [Google Scholar] [CrossRef] [Green Version]
- Yueh, S.C.H.; Lai, Y.A.; Chen, W.L.; Hsu, H.H.; Mao, S.J.T. An improved method for haptoglobin 1-1, 2-1, and 2-2 purification using monoclonal antibody affinity chromatography in the presence of sodium dodecyl sulfate. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 845, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Huang, Y.; Zhang, Y.; Meng, Q.; Luo, J.; Fan, B.; Ma, G.; Su, Z. A simple and rapid procedure for purification of haptoglobin from human plasma fraction IV. Artif. Cells Blood Substit. Immobil. Biotechnol. 2011, 39, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.S.; Palmer, A.F. Tangential flow filtration of haptoglobin. Biotechnol. Prog. 2020, 36, e3010. [Google Scholar] [CrossRef] [PubMed]
- Schaer, C.A.; Owczarek, C.; Deuel, J.W.; Schauer, S.; Baek, J.H.; Yalamanoglu, A.; Hardy, M.P.; Scotney, P.D.; Schmidt, P.M.; Pelzing, M.; et al. Phenotype-specific recombinant haptoglobin polymers co-expressed with C1r-like protein as optimized hemoglobin-binding therapeutics. BMC Biotechnol. 2018, 18, 15. [Google Scholar] [CrossRef] [PubMed]
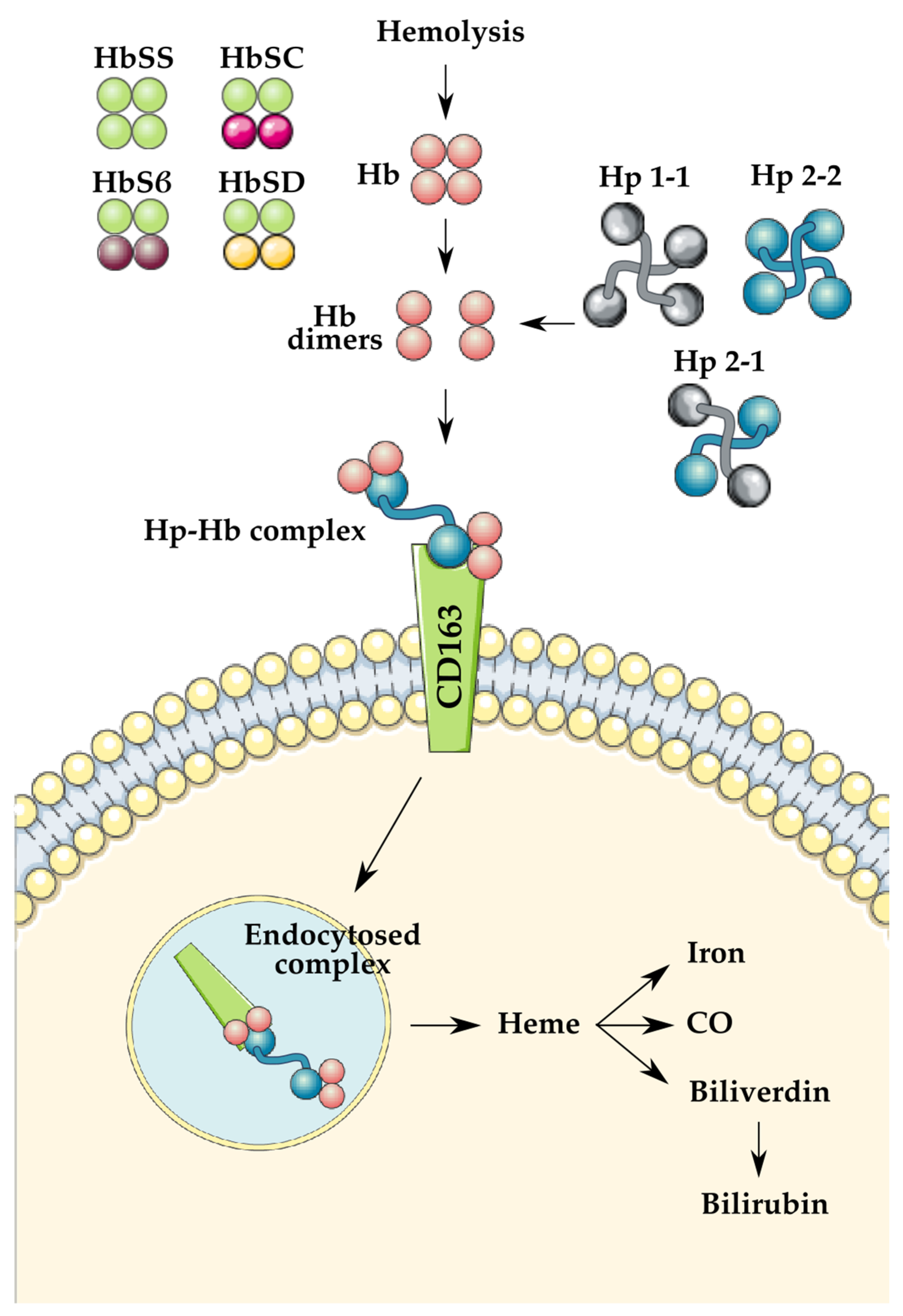


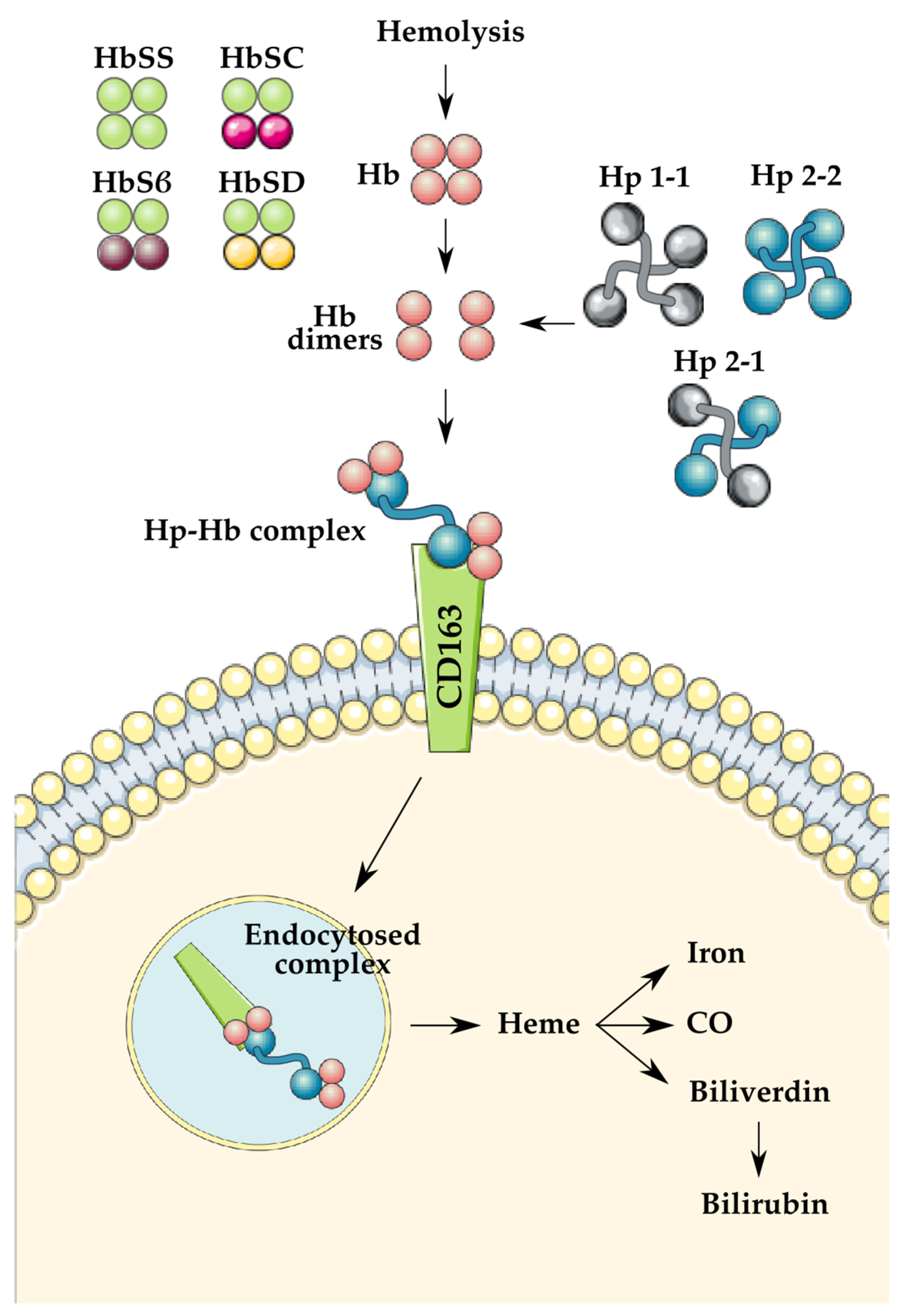{kind=link}
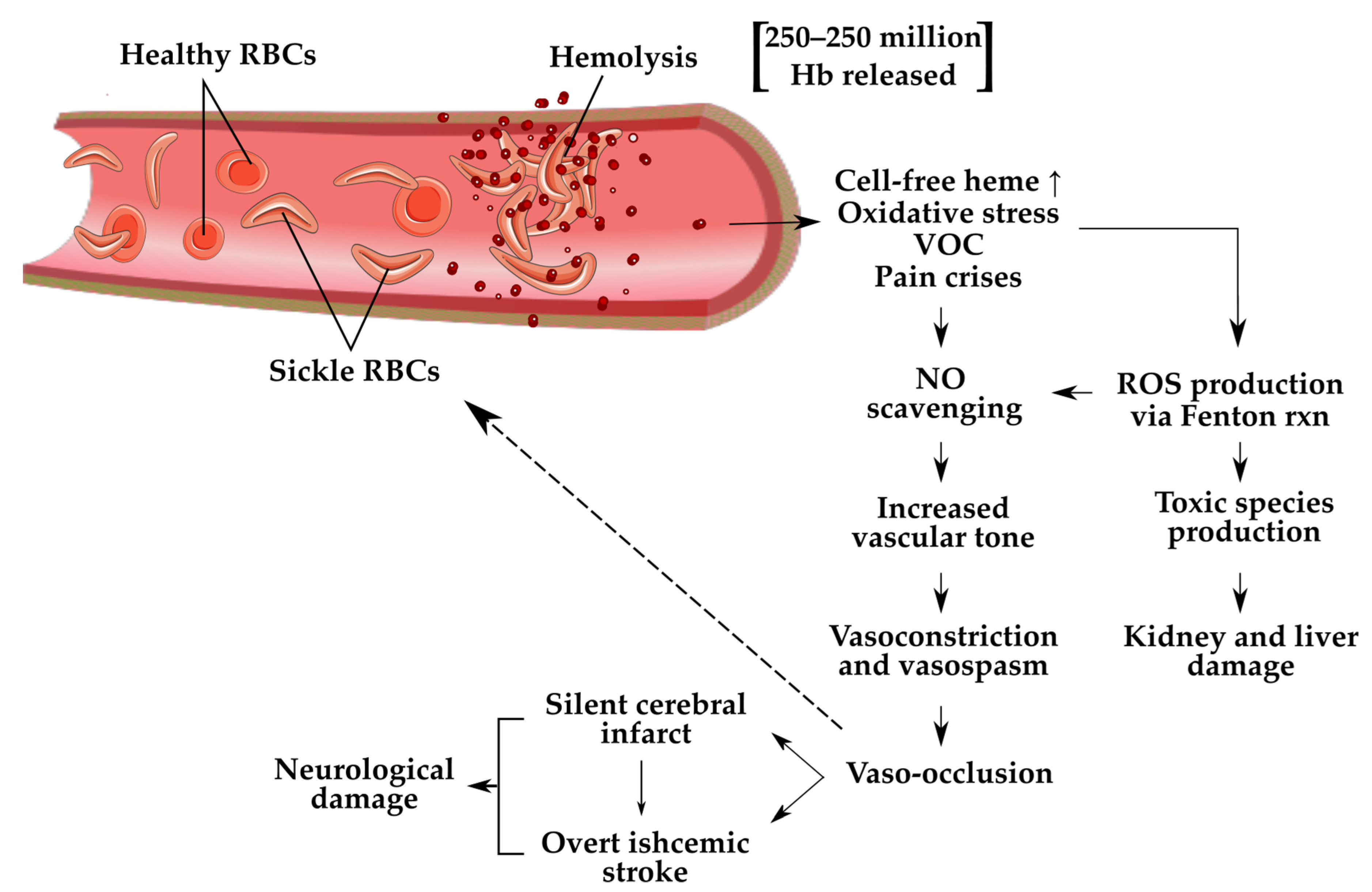{kind=link}
| Reference | Descriptive Parameters | Study Design | Anatomical Outcomes | Functional Outcomes |
|---|---|---|---|---|
| Atkinson et al. [9] Seasonal childhood anaemia in West Africa is associated with the haptoglobin 2-2 genotype | 671 Mandinka + Fulani children of age range 2–6 years No chronic illness Jul 2001–Jan 2002 West Kiang, Gambia | Prospective cohort study | - No significant trend observed between the Hp genotype and any sickle cell genotype - Hp genotype was not associated with baseline Hb levels, but children carrying Hp 2-2 showed twice the decrease in Hb over malaria season (p = 0.045) as Hp 1-2 children - Glucose-6-phosphate dehydrogenase (G6PD) deficiency was not significantly correlated with Hb drop in children with SCD | - Hp 2-2 was associated with a greater risk for anemia in malaria-infected children |
| Adekile and Haider [28] Haptoglobin gene polymorphisms in sickle cell disease patients with different βS-globin gene haplotypes | 82 Kuwaiti patients with SCD and 49 Kuwaiti control patients 54 South Nigerian patients with SCD and 32 South Nigerian control patients | Prospective case–control study | - Insignificant distribution pattern of Hp genotypes (p = 0.78, p = 0.41) - Significant (χ² = 31.4, p < 0.01) Hp genotype distribution between SCD groups - 52.4% of Kuwaiti patients with SCD were Hp 2-2 and only 16.7% of South Nigerian patients with SCD were Hp 2-2 - 37.5% of South Nigerian patients with SCD were Hp 1-1 and only 4.9% of Kuwaiti patients with SCD were Hp 1-1 - No significant difference among South Nigerian states - Kuwaiti patients with SCD and controls showed almost no difference in HP 2 allele distribution (73.8%, 71.4%) - South Nigerian patients with SCD and controls showed almost no difference in HP 1 allele distribution (60.7%, 54.7%) - No significant difference (p = 0.29) was found across Hp allele frequency studywide | - No significant differences were found between HP 2 allele presence and VOC frequency in the Kuwaiti group - Kuwaiti patients with SCD showed frequent VOC and no HP 2 allele association was found (Nigerian patients not stratified) - Stroke was uncommon among the Kuwaiti patients with SCD |
| Cox et al. [29] Haptoglobin, α-thalassaemia and glucose-6-phosphate dehydrogenase polymorphisms and risk of abnormal transcranial Doppler among patients with sickle cell anaemia in Tanzania | 601 Tanzanian patients with SCD <24 years 2004–2005, 2009 and 2010 Excluded blood transfusion in past 2 months, a sickle crisis in the past 2 weeks, history of stroke | Cross-sectional descriptive study | - Of the 601 patients with SCD, 23.46% had Hp 1-1, 56.64% had Hp 1-2, and 19.91% had Hp 2-2 genotypes - No significant difference between Hp 1-1 and Hp 2-2 in terms of CBF (p = 0.633) - 2-deletion α-thalassemia was inversely associated with CBF (p = 0.002) - Hb levels were inversely associated with CBF (p = 0.007) | N/D |
| Barbosa et al. [8] Haptoglobin and myeloperoxidase (-G463A) gene polymorphisms in Brazilian sickle cell patients with and without secondary iron overload | 78 Brazilian patients with homozygous SCA (HbSS) (34 M, 44 F), aged 21–65 years, 59 patients acquired iron overload and 19 patients normal iron, Federal District, Brazil | Cross-sectional study (convenience sample) | - 32% of patients with SCA with acquired iron overload exhibited Hp 2-2 - Hp 1F-1S and Hp 1F-1F were observed in only 15% of the 78 patients with SCA - Hp 2-2 patients with iron overload had a higher platelet count, but those without iron overload presented inconsistencies - No statistically significant distribution of Hp genotypes between iron overload and normal patients with SCA - Patients with iron overload and Hp 2-2 might experience a lesser change in blood flow dynamics due to SCA | - Lower risk of stroke linked to less Hp 1S-2 frequency than predicted (p = 0.005) - Hp 1S-2 showed the highest percentage of hospitalization for stroke and sequelae of stroke - Hp 1S-2 showed the highest hospitalization and sequelae of stroke, according to a large deviation from HWE; 21.57 patients predicted, only 10 patients observed - Hp 1F-1F group presented no patients with hospitalization for stroke - Hp 1S-2 and stroke hospitalization showed a significantly high odds ratio (OR = 6.346, p = 0.005) similar to Hp 1S-2 and sequelae of stroke (OR= 6.556, p = 0.005) |
| Olatunya et al. [26] Haptoglobin gene polymorphism in patients with sickle cell anemia: findings from a Nigerian cohort study | 101 Nigerian patients with stable SCA (67 M, 34 F) of age range 2–21 years, median 9 years SCA patients no crises for 1 mo and no transfusion for 100 days 64 healthy control patients; 40 M,24 F Ado Ekiti, Nigeria | Cross-sectional descriptive study | - No significant genotype distribution found between SCA and control groups - The HP 1 allele was more frequent among patients with SCA (~62%) but less frequent than controls (~73%); it was not statistically different (p = 0.06) - Study supports previous northeast Brazilian and Nigerian studies where the phenotype was more severe; genotypes associated with the HP 1 allele were more common among Nigerian patients with SCA | - Stroke identified in 5 patients: 2 Hp 1-1, 1 Hp 2-1, 2 Hp 2-2. Insignificant distribution (p = 0.375) - Study contrasts previous studies suggesting that certain SCA patient Hp genotypes (Hp 2-2) influence poor clinical health - Hp genotype is likely not a reliable indicator of clinical health in patients with SCA due to various insignificant results produced by this study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edwards, O.; Burris, A.; Lua, J.; Wilkie, D.J.; Ezenwa, M.O.; Doré, S. Influence of Haptoglobin Polymorphism on Stroke in Sickle Cell Disease Patients. Genes 2022, 13, 144. https://doi.org/10.3390/genes13010144
Edwards O, Burris A, Lua J, Wilkie DJ, Ezenwa MO, Doré S. Influence of Haptoglobin Polymorphism on Stroke in Sickle Cell Disease Patients. Genes. 2022; 13(1):144. https://doi.org/10.3390/genes13010144
Chicago/Turabian StyleEdwards, Olivia, Alicia Burris, Josh Lua, Diana J. Wilkie, Miriam O. Ezenwa, and Sylvain Doré. 2022. "Influence of Haptoglobin Polymorphism on Stroke in Sickle Cell Disease Patients" Genes 13, no. 1: 144. https://doi.org/10.3390/genes13010144
APA StyleEdwards, O., Burris, A., Lua, J., Wilkie, D. J., Ezenwa, M. O., & Doré, S. (2022). Influence of Haptoglobin Polymorphism on Stroke in Sickle Cell Disease Patients. Genes, 13(1), 144. https://doi.org/10.3390/genes13010144