1. Introduction
Marfan syndrome was described in 1896. Ehlers–Danlos syndrome, although first observed by Hippocrates, was described in modern medicine in the early 1900s. Loeys–Dietz syndrome was first reported in 2005. These are “syndromic” manifestations of thoracic aortic aneurysm disease (TAA), in which other organ systems besides the aorta show clinically discernible features.
In 1997 and 1999, Milewicz and our Yale team independently described essentially identical family patterns of “non-syndromic” TAA, in which the aorta (with its central branches) was affected, without external stigmata of inherited disease [
1,
2].
In the interim, advances in molecular genetics, especially the advent of affordable Whole Exome Sequencing achieved in the years since the first publication of J. Craig Venter’s gene sequence by Celera in 2001, [
3] have permitted light-speed progress in identifying new causes of thoracic aortic disease. The contributions of Milewicz and her team have been monumental. This has led to at least 34 specific genes currently implicated in the causation of TAA. New causative genes are added every year. The sum total of causative genes is published in regular review articles in the journal AORTA [
4].
When a suspicious new variant at a specific allelic location is identified, it is classified as a “Variant of Uncertain Significance (VUS)”, based on the guidelines of the American College of Medical Genetics and Genomics [
5,
6]. The variant remains so classified until strong information accumulates of its being truly causative rather than an incidental finding. A natural friction is encountered between the geneticist (who wishes not to be proven wrong by prematurely “calling” a potentially innocuous gene) and the clinician (often a surgeon), who cannot wait 5, 10, or 15 years for more families to be uncovered to confirm unequivocally that the specific variant segregates with the disease phenotype (aneurysm or no aneurysm) in multiple family trees. The patient can easily be lost to aortic dissection while unassailable confirmation of causative status of a variant is pursued [
7].
Unlike many diseases, which receive small genetic input from multiple variants in many genes, TAA is special. Most genetically triggered TAAs (that are known to date) are caused by a single nucleotide polymorphism (a variation in a single base in a DNA sequence) in a single gene [
8,
9].
There are several general methods to pursue initial analysis of the potential significance of a VUS:
Frequency in the general population. The rarity of the variant is important. For TAA, a rather unlikely disease, the variant has to be rare, certainly less than 1/10,000 human beings (since according to epidemiologic studies, TAA is estimated to occur in 10 out of 100,000 individuals in the general population per year). If the variant is not rarer than that level, it is not of causative interest (unless we suspect a recessive type of TAA, which is very rare), because the variant is more common than the disease itself. Large databases are now available that include maps of more than 140,000 individuals (such as the Genome Aggregation Database (gnomAD) [
10]).
In silico analysis. Computerized methods can predict the molecular impact of a specific variant at a specific spot in one particular gene. Some variants may be of almost no impact. Others can stop cold the reading of the gene beyond the variant, with disastrous protein manufacturing consequences. A dramatic negative effect on reading of the gene suggests the variant in question is likely disease-causing.
Conservation in phylogeny. The degree of conservation of the normal allele at one specific location in one gene is evidence of its importance. The further down the phylogenetic tree that the conservation of the allele goes, the more important is that location on that gene. Computerized methods are available for exact determination of degree of conservation of the normal variant at any specific location (from human to monkey, pig, dog, rat, mouse, fish). Conservation of a specific variant in phylogeny is among the strongest predictors of its importance in causation of disease.
Animal models. It is possible to insert the variant allele into an animal embryo and monitor for disease development. This has generally been done in mammals (mice) and is very time-consuming and expensive [
11].
Familial Variant Segregation Analysis. Other affected and unaffected members of the same family can be tested for the VUS carrier status in order to determine whether the VUS segregates with the disease or not. Of course, issues of (1) degree of penetrance (the likelihood that a carrier manifests the disease) and (2) duration of time after birth required for an aortic aneurysm to manifest (usually decades) need to be taken in to account when conducting familial segregation analysis.
In this report, we describe a novel approach for efficient clinical analysis of significance of TAA variants via inducing genetically mutant zebrafish. Experiments in zebrafish are generally inexpensive and highly reproducible and yield rapid results because of the quick maturation of the species. Furthermore, the translucent nature of the zebrafish renders gross and microscopic analysis of internal organs more facile than in other species.
We reasoned that if the zebrafish method was feasible and informative, this model could provide a means for rapid assessment of the significance of a mutation and permit rapid introduction of that mutation into clinical decision making. Even if the precise clinical mutations cannot be recreated in the zebrafish in a rapid fashion, the associated genes can be rapidly screened for potential significance in a matter of weeks or months by inactivating them and studying zebrafish larval phenotypes. If successful, this rapid approach could be of life-saving significance in the TAA population—via earlier recognition of disease causation, with consequently expedited treatment.
2. Materials and Methods
We examined the impact of inducing loss of function mutations in genes potentially implicated in thoracic aortic aneurysm disease on anatomy and clinical outcome in zebrafish. We also examined the impact on zebrafish of mutations in some genes known to be implicated in Marfan’s disease (fbn1) and Ehlers–Danlos syndrome (col1a2, col5a1, and col5a2a/b). We used these known disease related genes as “positive controls”, which should be reflected morphologically if the zebrafish model were effective as an evaluative tool.
General zebrafish maturation and care methodology. Adult AB zebrafish were maintained on a standard 14 h/10 h dark/light cycle in 3.0 L housing with constant water flow (Iwaki Aquatic, Holliston, MA, USA). Adults were fed a standard lab diet of Gemma Micro 500 and brine shrimp, once daily (Skretting, Stavanger, Norway). Zebrafish embryos were obtained by natural matings and maintained in E3 medium (5 mM NaCl, 160 µM KCl, 333 µM CaCl
2, 416 µM MgCl
2) at 28.5 °C. All anesthesia was carried out in 0.16 mg/mL MS-222 (Syndel, Ferndale, WA, USA). Care protocols were modeled after standard methods at Boston Children’s Hospital [
12,
13]. All animal care and handling were carried out under the approval of IACUC (Protocol #: 2019-20274).
Validation of CRISPR reagents and generation of zebrafish transient mutants. For each of the eight genes analyzed in this study, two sequence-specific crRNAs were designed using the CRISPOR web tool (
Table 1) [
14]. These were annealed to tracRNA to form an RNA duplex stock (33 µM) which was then complexed with 10 µg/µL Alt-R-
S.p. Cas9 Nuclease V3 (IDT) in Cas9 buffer (20 mM HEPES, 150 mM KCl, pH 7.5) at a 1:1 molar ratio according to the manufacturer’s instructions. This was then supplemented with sterile 2% phenol red to a final concentration of 0.2% phenol red containing a roughly 3.3 µM concentration of each CRISPR/Cas9 ribonucleoprotein complex. This constituted the injection mixture.
Zebrafish embryos were obtained at the 1–2 cell stage and 1 nL of the described mixture was injected into each embryo. Embryos were allowed to recover in E3 medium. ~10 embryos were collected at 1–3 days post fertilization (dpf) and their DNA was harvested by boiling them in 50 µL of 100 mM NaOH for 20 min. The pH of the resulting solution was adjusted by adding 20 µL 1M Tris-HCl pH 7.5.
Teleosts, of which zebrafish are a member, underwent a whole genome duplication event approximately 350 million years ago [
15]. Consequently, they often have redundant paralogues of mammalian genes which necessitates inactivating both copies. This was the case for both
emilin1 and
col5a2, which each have two paralogues
a and
b in zebrafish. We developed two gRNAs for each paralogue and injected a mixture containing all four gRNAs with Cas9 in an attempt to exclude any compensatory function. This method has been successfully applied for other diseases, including phenotypic screening of genes involved in spinal cord injury or defective locomotor behaviors [
16,
17,
18].
These DNA preparations were subjected to site-specific PCR (
Table 1) to detect heteroduplex DNA generated by CRISPR/Cas9 indels. The same PCR amplicons were isolated by TOPO cloning and directly sequenced to assess the efficacy of each gRNA individually.
Briefly, a 300–800 bp amplicon was generated at each targeted locus in both targeted and non-targeted (i.e., unmodified) fish. These were then denatured and reannealed using an extended 0.2 °C/s ramp. The resulting DNA was digested with T7 endonuclease (NEB) and run out on 2% agarose gel for analysis. If a given RNP was determined to be effective, it was used further for analysis of F0 zebrafish. As an additional confirmation of this approach, we cloned and sequenced individual target molecules to assess the percent efficacy of each gRNA used in this approach. Study-wide indel efficiency was 86%; for most gRNAs we were unable to recover non-mutant sequences from individual clones taken from injected fish, suggesting a very high degree of mutation (
Figure 1).
Brightfield microscopy. Injected fish were analyzed between 1–6 dpf for potential phenotypes using an Olympus MVX-10 Macro Zoom microscope. Findings searched included: cerebral hemorrhage, aortic hemorrhage, scoliosis/axial curvature, and abnormalities of cardiac and vascular morphology (dorsal aorta (DA) and intersegmental vessels (INV)).
Confocal microscopy. In the case of emilin1a/b and col5a1 transients, we analyzed Tg(kdr:GFP)zn1 and Tg(kdrl:ras-mCherry; fli1a:nls-GFP)s896/y7 transgenic lines using a Leica SP8 confocal microscope to identify phenotypes not visible under brightfield. Here, we assessed the diameter and endothelial cell density of the dorsal aorta (DA) and intersegmental vessels (ISVs) of the trunk.
3. Results
3.1. Zebrafish Modeling
We examined the impact of the loss of function of multiple genes on anatomy and clinical outcome in zebrafish.
Specifically, we examined two genes which have recently arisen in our clinical practice:
EMILIN1 and
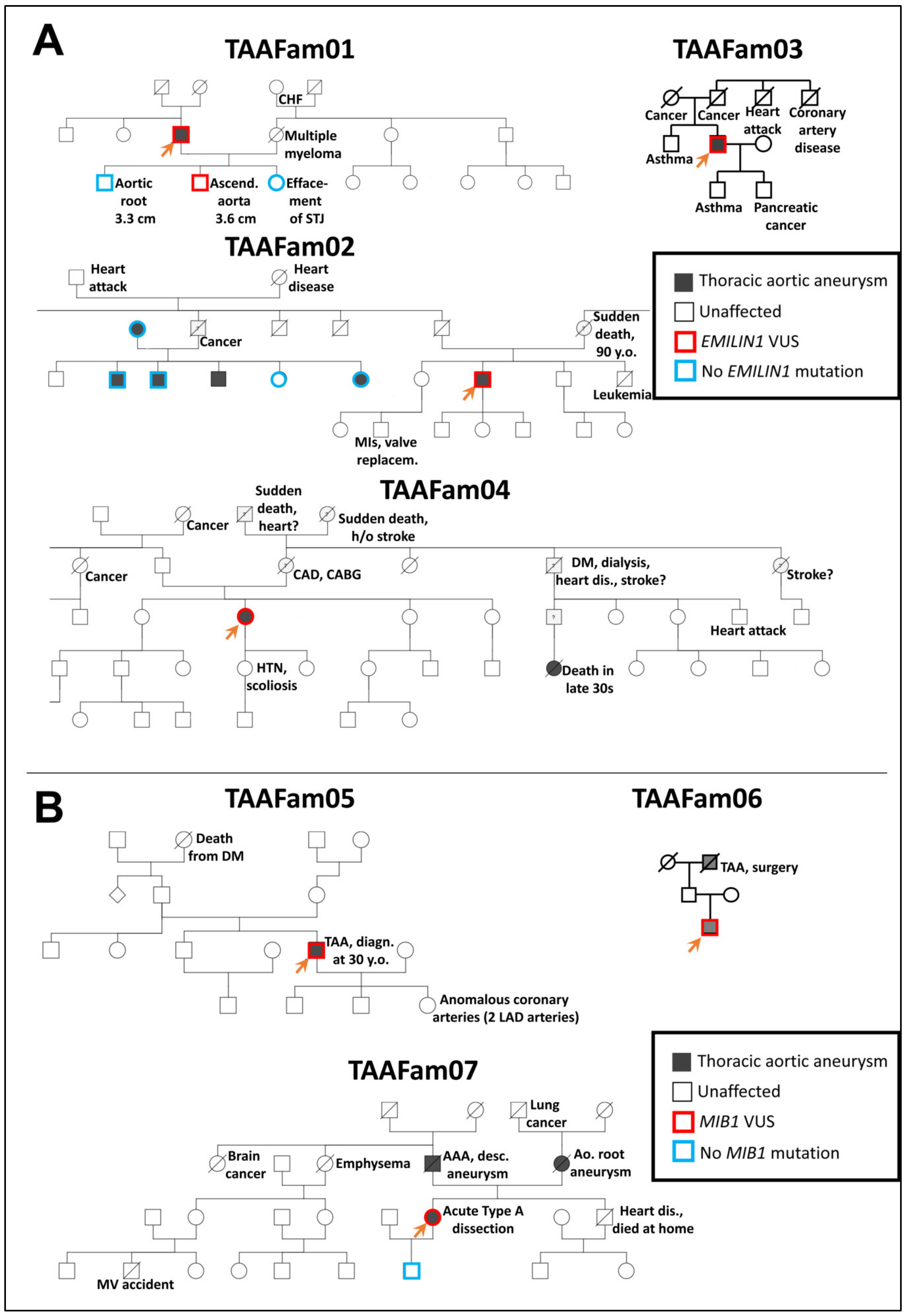
MIB1. We chose these two genes because they have presented mutations in our clinical practice in patients with thoracic aortic disease. It must be emphasized here that due to technical obstacles, we are not recreating the exact mutations in zebrafish as we observe clinically—rather, we are generating loss-of-function alleles in the associated genes. The pertinent clinical pedigrees in these families are shown in
Figure 2.
The
EMILIN1 (or, elastin microfibril interface 1) gene is an extracellular matrix glycoprotein abundantly expressed in elastin-rich tissues (such as the blood vessels, skin, heart, and lung) and primarily localized at sites where elastin and microfibrils are in proximity [
19]. Defects in
EMILIN1 expression adversely affect lamellar structure and function and increase aortic wall inflammation and have been associated with connective tissue disease [
19,
20,
21,
22]).
The
MIB1 gene (“Mindbomb” gene) encodes the E3 ubiquitin-protein ligase MIB1, which regulates endocytosis of Notch ligands [
23] and has been associated with left ventricular non-compaction syndrome (OMIM 615092) [
24].
At the earliest stages of zebrafish development (1–2 days post fertilization-dpf) the zebrafish dorsal aorta is comprised of a single-celled endothelial tube rather than the multilayered structure that is characteristic of the mature mammalian aorta [
25,
26]). Although zebrafish embryos will progressively add mural cells to develop a multilayered aorta during the late larval and juvenile developmental periods [
25], the aorta initially resembles mammalian capillaries in structure. Consequently, attempts to model TAA in the zebrafish embryo must be both careful and broad in scope.
We were able to identify several abnormal anatomic zebrafish characteristics in our “positive controls” and in our novel variants needing evaluation for deleterious effects, namely emilin1 and mib1.
These abnormal characteristics are not seen in normal zebrafish not subjected to mutagenesis of deleterious genes. As depicted in
Figure 3,
Figure 4 and
Figure 5, these aberrant characteristics include (in various combinations for the deleterious genes):
Peri-aortic hemorrhage (manifested as red splotching adjacent to the aorta).
Cranial hemorrhage (manifested as red splotching in the brain).
Scoliosis (manifested as curvature of the spine upward toward the head, at times producing marked structural deformity of the entire body of the zebrafish).
Reduced diameter of the dorsal aorta (DA) and the intersegmental vessels (ISVx) (similar to mouse phenotype [
22,
23,
24,
25,
26,
27]) and “drop out” interruption of the DA.
Lower cell counts in the dorsal aorta and the intersegmental vessels
Cardiomegaly (or, more properly, distention of the pericardial sac, at times severe).
3.2. Genetic Modifications
Specific details for each genetic modification of the zebrafish are supplied below. The frequency of observed abnormal manifestations varied for each targeted gene, but each target displayed some degree of phenotypic abnormality (
Figure 1F–G).
3.2.1. Fibrillin 1 and Collagen Subunits
We initially addressed a suite of genes previously implicated in human cases and animal models of TAA [
4]. These are the “positive controls” for our study, designed to show that the zebrafish model can detect adverse consequences from known pathogenic genes.
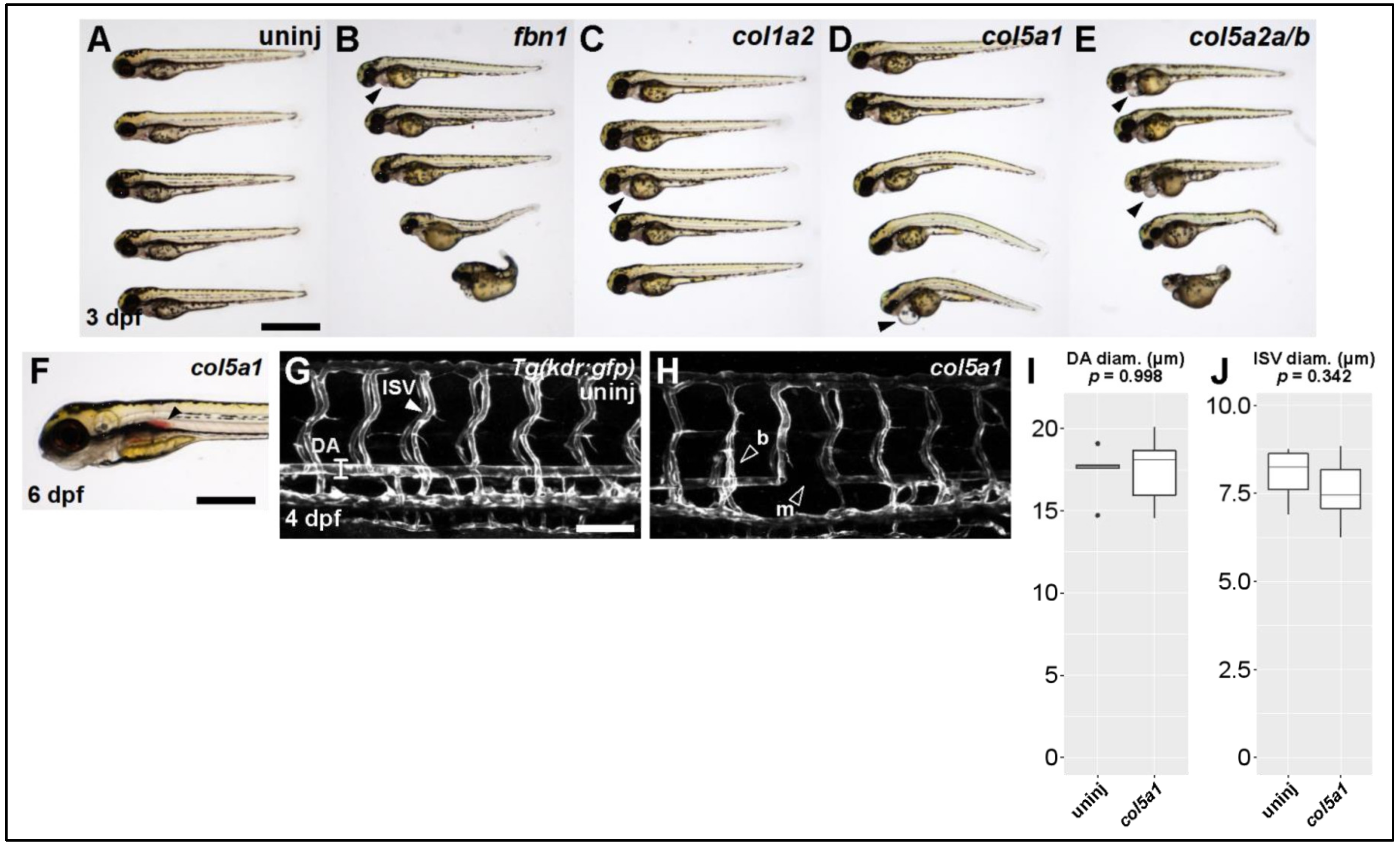
We injected CRISPR/Cas9 against
fbn1 (Marfan gene) and the collagen genes
col1a2,
col5a1, and
col5a2a/b (Ehlers–Danlos genes). The most pronounced defect across all conditions was some degree of cardiac edema or cardiomegaly (
Figure 3A–E, filled arrowheads). Scoliotic or kyphotic phenotypes were also frequently observed across all conditions. In a small subset of targeted fish, we observed hemorrhages of the dorsal aorta (
Figure 3F, filled arrowhead)—a phenotype we never observe in control fish and which seems directly comparable to rupture or dissection of human TAA.
We proceeded to collect quantitative data on the vasculature of the trunk in
col5a1 fish. Using the
Tg(kdr:gfp) fish line, which labels all vascular endothelial cells, we were able to visualize and measure both the dorsal aorta (DA) and the intersegmental vessels (ISVs) of the trunk (
Figure 3G,H). Although we did not observe statistically significant differences in the diameter of these vessels (
Figure 3I,J), we did observe missing vascular segments in both DA and ISVs as well as inappropriate vascular branching in ISVs (
Figure 3H, empty arrowheads). Taken together, these data suggest that knocking out known TAA-associated genes produces abnormal vascular phenotypes in larval zebrafish as young as 3 dpf.
3.2.2. Mindbomb 1
Mindbomb 1.
Mib1 is a known E3 Ubiquitin ligase that regulates Notch signaling. It is well known in zebrafish to regulate both angiogenesis and neurogenesis [
28,
29,
30]. We designed CRISPR/Cas9 RNPs targeting the same locations as the clinically identified variants of unknown significance (
Figure 4A) and then validated these using a standard T7 endonuclease assay to verify that they were generating indel variants at those locations (
Figure 4B).
Injection of
mib1 CRISPR/Cas9 generated extremely severe phenotypes. Axial curvature was massively disrupted (
Figure 4C,D). We also routinely observed hemorrhaging, both in the cerebral vasculature and in the aorta (
Figure 4E–G). This rate of hemorrhage was significantly greater in
mib1-deficient fish when compared to very rare events in controls (
Figure 4H, chi-square test,
p < 0.001).
3.2.3. Emilin 1a and 1b
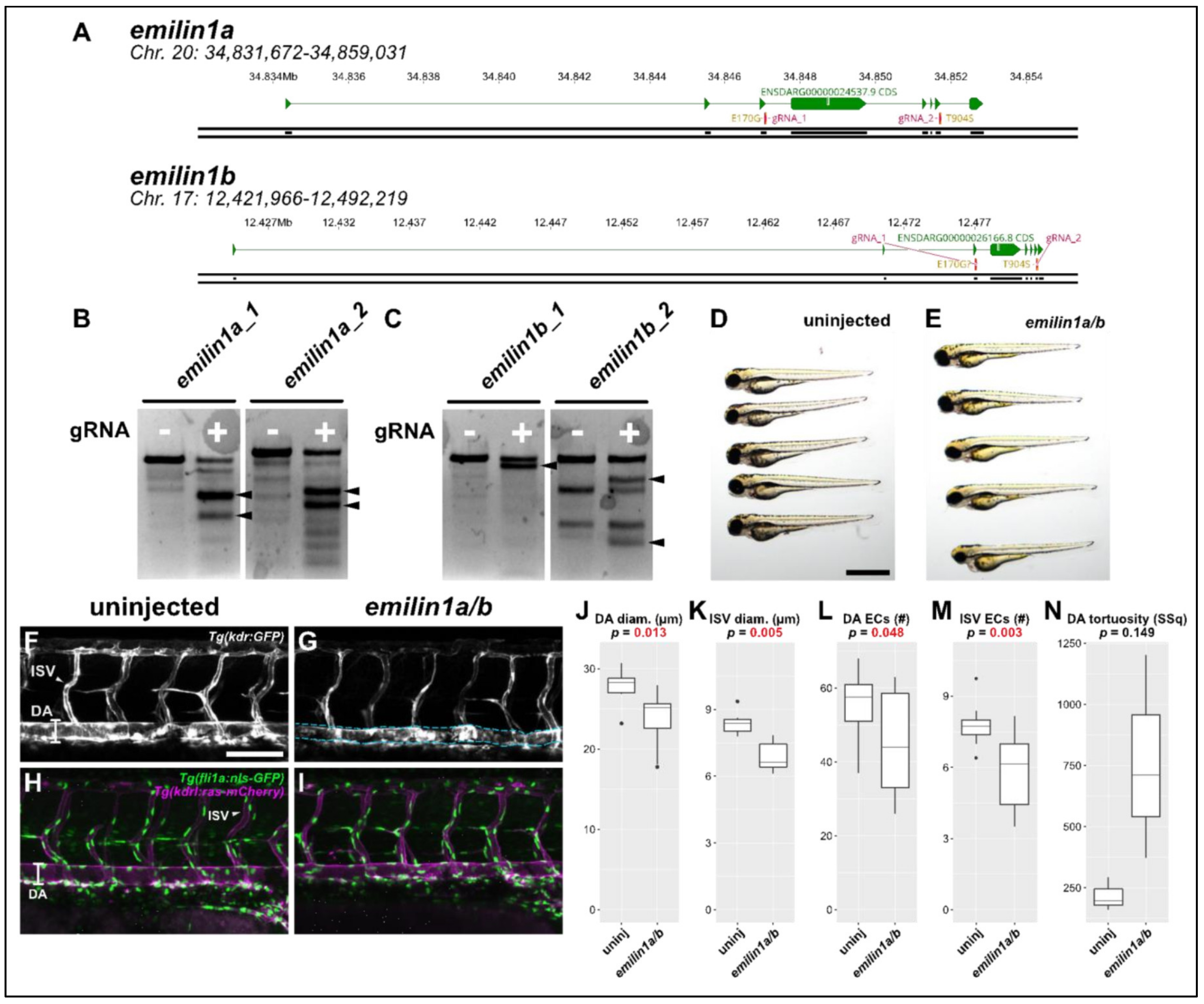
Elastin microfibril interfacer 1 (Emilin1) is an extracellular matrix protein that associates with microfibrils and elastin; it is therefore a compelling candidate for a gene implicated in TAA, which relies on the proper development and maintenance of elastic tissues such as those found in large-diameter blood vessels. We again generated CRISPR/Cas9 targeting the sites of each of the observed patient variants (
Figure 5A) and validated these reagents as before (
Figure 5B,C). Initially, we found no gross phenotypic abnormalities in
emilin1a/b knockout fish (
Figure 5D,E). Consequently, we proceeded to do a more careful analysis of the zebrafish trunk vasculature.
Morphological analysis of the
emilin1a/b knockout fish revealed that both DA and ISVs exhibited significantly smaller diameters. This phenotypic profile is entirely consistent with previous reports in mouse which revealed the same findings [
22,
27]. We subsequently performed cell counts in the endothelial cells of these vessels using the
Tg(fli1a:nls-gfp) line to determine whether this reduced size was due to fewer endothelial cells being present. In both vessel types, endothelial cell number was substantially reduced (
Figure 5L,M). A final analysis of the DA to assess its tortuosity (
Figure 5G, blue dashed lines) is suggestive of an overall trend towards greater sinuousness but ultimately was not statistically significant.
4. Discussion
Zebrafish have substantial features that make them very suitable for genetic investigations of the cardiovascular system and aorta. They have rapid gestation and maturation. They are transparent, so that the heart and aorta can be easily visualized by direct inspection. They can be examined by light microscopic videography. Medications can be administered by simple instillation into the bath water, from which they are systemically absorbed through the gills or skin. In short, zebrafish provide a valuable tool for study of aortic pathologies associated with specific genetic mutations.
Zebrafish have been used in non-clinical laboratory studies of various aspects of aortic disease. Abrial and colleagues at Massachusetts General Hospital have used zebrafish to study post-injury myocardial regeneration pathways as well as development of the aortic arch vessels during embryogenesis. They engineered zebrafish lacking two genes in the arch development pathway; surprisingly, those zebrafish also within days developed a Marfan’s-like phenotype, including aortic aneurysm [
31]. Ton and colleagues studied a knockdown of a
Col22a1 gene in zebrafish, finding a 2.5-fold increase in the number of cerebral hemorrhages compared to the wild type zebrafish, which they attributed to abnormal vessel wall permeability [
32]. They also noted associated vessel dilatation, irregular vessel contour, and irregular vascular wall structure. They found that the frequency of hemorrhages increased with heat stress (33.5 °C), forced rapid swimming, or monitoring to older zebrafish age (up to 2 years) [
32]. In their studies of the toxicity of cigarette products on the cardiovascular system of zebrafish, Folkesson and colleagues found distinct evidence of toxicity to the vascular tree and aorta, manifested as cerebral hemorrhages and/or aortic dilatation [
33]. Dietz and Milewicz also found zebrafish models of aortic disease useful in their basic science studies of aortic aneurysm and bicuspid valve disease [
34,
35]. In addition to the zebrafish aneurysm related characteristics that we observed, some authors have noted pericardial edema, maldevelopment of the aortic arches, and major developmental abnormalities of the body of the zebrafish [
33].
In the present paper, we present our initial experience using zebrafish mutants with an eye toward permitting rapid, low cost, clinical estimation of the clinical significance of mutations encountered in the course of standard clinical aortic care.
We feel that this initial experience with evaluation of potential aortic aneurysm causative genetic variants by targeted genes in the zebrafish is encouraging. A zebrafish “aortic phenotype” is emerging through these initial investigations in the zebrafish mutant models. This involves hemorrhage (peri-aortic or cerebral), cardiomegaly, scoliosis, decreased vessel caliber, and decrease in number of vascular cells. This phenotype is certain to vary in zebrafish by gene, just as in humans the different genetically mediated syndromes have different manifestations.
The research reported here has several addressable limitations. First, our initial approach does not precisely duplicate the mutations observed in human patients, which are often missense mutations involving single amino acid substitutions. Rather, we generate nonsense loss-of-function mutations at the same loci as the observed patient mutation. In essence, we are really investigating the involved gene rather than the precise clinical variant itself. However, this approach is still conceptually useful as we believe many of the human mutations are indeed loss-of-function alleles based on in silico prediction; however, it is fair to say that our fish models are not one-to-one duplicates of the observed human mutations. Furthermore, if the native variant produces a nonsense or stop codon, our current work is equivalent and properly representative of the impact of the variant.
This, our initial study, may, thus, exaggerate the impact of the human mutation. To address this limitation, we will expand our study by attempting to rescue these loss-of-function alleles using injected mRNA cloned from patients carrying the exact mutation we wish to model. A functioning variant mRNA will rescue phenotypic defects. Conversely, a human mutation affecting protein functionality will be unable to rescue the zebrafish mutants.
A second limitation lies with the timing of our phenotypic characterization, which is restricted to the embryonic and larval stages. We studied only F0 zebrafish. Extending experimental time would permit assessment of F1 and F2 generations, thus minimizing the potential for persistence of wt fish in the F0 generation. We plan to investigate our VUS mutant phenotypes longer into the zebrafish lifecycle. This likely will permit identification of additional pathophysiological consequences beyond the TAA-related defects associated with embryonic malformation.
Additionally, the COL5A1 and COL5A2 genes chosen as positive controls represent classical Ehlers–Danlos syndrome, which is not strongly aortic but more soft tissue oriented. We plan in future experiments to test additional, more strongly aortic genes. However, even with the COL genes, we did find both aortic and skeletal abnormalities in the present studies—reflecting high discriminatory abilities of our model. As well, we did include the strongly aortic FBN1 gene even in these early studies, which proved a strong positive control.
Additionally, Marfan’s disease is known to be related, in many cases, to dominant negative activity of the mutant protein. Our modeling may not be reflective of such mechanisms. In the future the ability to investigate more VUSs in more TAA genes will build a phenotypic map of TAA associated VUSs that will enhance potential prediction of variant casualty. Furthermore, this approach may well expand our understanding of cellular processes associated with TAA-related anomalies. Furthermore, zebrafish VUS mutant models may enable screening for drugs or small molecules that can revert the course of these aberrant phenotypes, potentially contributing to future personalized therapeutics for the treatment of TAA. Even though this initial study has limitations, it does provide a novel opportunity for enhancing our biological and clinical understanding of TAA disease.
Our data suggest that the zebrafish mutagenesis model can evolve into a useful tool for rapid evaluation of novel genetic variants of unknown significance. If this eventuates, this is likely to contribute substantially to patient care. Specifically, a near immediate provisional answer as to “disease causing” versus “benign” variants may be facilitated. This would represent a considerable advance compared to the current status of waiting, possibly decades, for co-segregation of variant and phenotype to be demonstrated unequivocally in human families. The potential for lethal clinical events to occur in family members during these long “limbo” periods in the traditional clinical evaluation may be mitigated by early confirmation of disease induction in the zebrafish model. Of course, we would recommend that the zebrafish findings be considered only as one more avenue of variant evaluation—taken together with the modalities discussed in the introduction of this paper—namely, frequency of the variation in the general population, preservation in phylogeny, in silico analysis, and segregation of disease with the variant in clinical analysis of large families.
Author Contributions
Conceptualization, J.A.E., A.P., S.N.; methodology, A.P., S.N.; formal analysis, A.P., S.N., D.P., B.A.Z.; Investigation, A.P., S.N., J.A.E.; resources, S.N., J.A.E.; data curation, A.P., D.P., M.A.Z.; writing—original draft reparation, J.A.E., A.P., S.N.; writing—review and editing, J.A.E., A.P., S.N., S.M., B.A.Z., M.A.Z.; visualization, A.P., B.A.Z., M.A.Z., J.A.E.; supervision, J.A.E., S.N., S.M.; project administration, J.A.E., S.N., S.M.; funding acquisition, J.A.E. All authors have read and agreed to the published version of the manuscript.
Funding
Intramural: Aortic Institute at Yale-New Haven (internal funds).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of Yale University School of Medicine (Protocol #: 2019-20274).
Informed Consent Statement
Not applicable.
Data Availability Statement
Available on request.
Conflicts of Interest
Elefteriades—Prinicpal, CoolSpine; Data/Safety Monitoring Board, Terumo; Consultant, CryoLife. None are relevant to this manuscript.
References
- Biddinger, A.; Rocklin, M.; Coselli, J.; Milewicz, D.M. Familial thoracic aortic dilatations and dissections: A case control study. J. Vasc. Surg. 1997, 25, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Coady, M.; Davies, R.; Roberts, M. Familial Patterns of Thoracic Aortic Aneurysms. ACC Curr. J. Rev. 2005, 14, 19. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Vinholo, T.F.; Brownstein, A.J.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2019 Update and Clinical Implications. AORTA 2019, 7, 99–107. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Elefteriades, J.A. Beating a Sudden Killer. Sci. Am. 2005, 293, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, B.A.; Bailey, A.E.; Coons, C.; Dykas, D.; Charilaou, P.; Tanriverdi, L.H.; Liu, L.; Tranquilli, M.; Bale, A.E.; Elefteriades, J.A. Routine Genetic Testing for Thoracic Aortic Aneurysm and Dissection in a Clinical Setting. Ann. Thorac. Surg. 2015, 100, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, C. New frontiers for zebrafish management. Methods Cell Biol. 2016, 135, 483–508. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, C. Advances in Zebrafish Husbandry and Management. Methods Cell Biol. 2011, 104, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, S.M.; Neuhauss, S.C. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genom. 2014, 289, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Kroll, F.; Powell, G.T.; Ghosh, M.; Gestri, G.; Antinucci, P.; Hearn, T.J.; Tunbak, H.; Lim, S.; Dennis, H.; Fernandez, J.M.; et al. A simple and effective F0 knockout method for rapid screening of behaviour and other complex phenotypes. eLife 2021, 10, e59683. [Google Scholar] [CrossRef]
- Keatinge, M.; Tsarouchas, T.M.; Munir, T.; Porter, N.J.; Larraz, J.; Gianni, D.; Tsai, H.-H.; Becker, C.G.; Lyons, D.A.; Becker, T. CRISPR gRNA phenotypic screening in zebrafish reveals pro-regenerative genes in spinal cord injury. PLoS Genet. 2021, 17, e1009515. [Google Scholar] [CrossRef]
- Hoshijima, K.; Jurynec, M.J.; Shaw, D.K.; Jacobi, A.M.; Behlke, M.A.; Grunwald, D.J. Highly Efficient CRISPR-Cas9-Based Methods for Generating Deletion Mutations and F0 Embryos that Lack Gene Function in Zebrafish. Dev. Cell 2019, 51, 645–657. [Google Scholar] [CrossRef]
- Doliana, R.; Canton, A.; Bucciotti, F.; Mongiat, M.; Bonaldo, P.; Colombatti, A. Structure, chromosomal localization, and promoter analysis of the human elastin mi-crofibril interfase located proteIN (EMILIN) gene. J. Biol. Chem. 2000, 275, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Rabajdova, M.; Urban, P.; Spakova, I.; Saksun, L.; Dudic, R.; Ostro, A.; Caprnda, M.; Kruzliak, P.; Adamek, M.; Marekova, M. The crucial role of emilin 1 gene expression during progression of tumor growth. J. Cancer Res. Clin. Oncol. 2016, 142, 2397–2402. [Google Scholar] [CrossRef]
- Capuano, A.; Bucciotti, F.; Farwell, K.D.; Davis, B.T.; Mroske, C.; Hulick, P.J.; Weissman, S.M.; Gao, Q.; Spessotto, P.; Colombatti, A.; et al. Diagnostic Exome Sequencing Identifies a Novel Gene, EMILIN1, Associated with Autosomal-Dominant Hereditary Connective Tissue Disease. Hum. Mutat. 2016, 37, 84–97. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, M.; Braghetta, P.; Sabatelli, P.; Mura, I.; Doliana, R.; Colombatti, A.; Volpin, D.; Bonaldo, P.; Bressan, G.M. EMILIN-1 Deficiency Induces Elastogenesis and Vascular Cell Defects. Mol. Cell. Biol. 2004, 24, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Kim, Y.-W.; Koo, B.-K.; Jeong, H.-W.; Yoon, M.-J.; Yoon, K.-J.; Jun, D.-J.; Im, S.-K.; Shin, J.; Kong, M.-P.; et al. Mind bomb 1 in the lymphopoietic niches is essential for T and marginal zone B cell development. J. Exp. Med. 2008, 205, 2525–2536. [Google Scholar] [CrossRef] [Green Version]
- Luxán, G.; Casanova, J.C.; Poveda, B.M.; Prados, B.; D’Amato, G.; MacGrogan, D.; Gonzalez-Rajal, A.; Dobarro, D.; Torroja, C.; Martinez, F.; et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 2013, 19, 193–201. [Google Scholar] [CrossRef]
- Stratman, A.N.; Pezoa, S.A.; Farrelly, O.M.; Castranova, D.; Dye, L.E., III; Butler, M.G.; Sidik, H.; Talbot, W.S.; Weinstein, B.M. Interactions between mural cells and endothelial cells stabilize the developing zebrafish dorsal aorta. Development 2017, 144, 115–127. [Google Scholar]
- Miano, J.M.; Georger, M.A.; Rich, A.; Bentley, K.L.D.M. Ultrastructure of Zebrafish Dorsal Aortic Cells. Zebrafish 2006, 3, 455–463. [Google Scholar] [CrossRef]
- Zacchigna, L.; Vecchione, C.; Notte, A.; Cordenonsi, M.; Dupont, S.; Maretto, S.; Ferrari, A.; Maffei, A.; Fabbro, C.; Braghetta, P. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell 2006, 124, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.M.; Schulte-Merker, S. How to Plumb a Pisces: Understanding Vascular Development and Disease Using Zebrafish Embryos. Dev. Cell 2017, 42, 567–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, M.; Kim, C.-H.; Palardy, G.; Oda, T.; Jiang, Y.-J.; Maust, D.; Yeo, S.-Y.; Lorick, K.; Wright, G.; Ariza-McNaughton, L.; et al. Mind Bomb Is a Ubiquitin Ligase that Is Essential for Efficient Activation of Notch Signaling by Delta. Dev. Cell 2003, 4, 67–82. [Google Scholar] [CrossRef] [Green Version]
- Schier, A.F.; Neuhauss, S.C.; Harvey, M.; Malicki, J.; Solnica-Krezel, L.; Stainier, D.Y.; Zwartkruis, F.; Abdelilah, S.; Stemple, D.L.; Rangini, Z.; et al. Mutations affecting the development of the embryonic zebrafish brain. Development 1996, 123, 165–178. [Google Scholar] [CrossRef]
- Abrial, M.; Paffet-Lugassy, N.; Jeffrey, S.; Jordan, D.; O’Loughlin, E.; Frederick, C.J., III; Burns, C.G.; Burns, C.E. TGF-beta Signaling Is Necessary and Sufficient for Pharyngeal Arch Artery An-gioblast Formation. Cell Rep. 2017, 20, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton, Q.V.; Leino, D.; Mowery, S.A.; Bredemeier, N.O.; Lafontant, P.J.; Lubert, A.; Gurung, S.; Farlow, J.; Foroud, T.M.; Broderick, J.; et al. Collagen COL22A1 maintains vascular stability and mutations in COL22A1 are potentially associated with intracranial aneurysms. Dis. Model. Mech. 2018, 11, dmm033654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkesson, M.; Sadowska, N.; Vikingsson, S.; Karlsson, M.; Carlhäll, C.-J.; Länne, T.; Wågsäter, D.; Jensen, L. Differences in cardiovascular toxicities associated with cigarette smoking and snuff use revealed using novel zebrafish models. Biol. Open 2016, 5, 970–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, R.A.; Aziz, H.; Woods, C.E.; Seman-Senderos, M.A.; Sparks, E.; Preuss, C.; Wünnemann, F.; Bedja, D.; Moats, C.R.; McClymont, S.A.; et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat. Genet. 2019, 51, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-C.; Gong, L.; Regalado, E.S.; Santos-Cortez, R.L.; Zhao, R.; Cai, B.; Veeraraghavan, S.; Prakash, S.K.; Johnson, R.J.; Muilenburg, A.; et al. MAT2A Mutations Predispose Individuals to Thoracic Aortic Aneurysms. Am. J. Hum. Genet. 2015, 96, 170–177. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Quantification of phenotypes and gRNA efficacy. (A) Amplicons for each gRNA target were generated by PCR and then TOPO-cloned to generate a library of targeted genomic DNA sequences. (B) Individual clones were amplified by colony PCR and sent for sequencing. (C) Sequences were aligned to wt to determine the incidence of indel mutations. (D) For most targets, we are unable to isolate wt DNA by cloning, indicating a very high degree of mutational efficiency (with the exception of emilin1b gRNA 2, which appears to be non-functional). (E) Quantification of cardiomegaly phenotypes for all genes at 2 and 5 dpf. (F) Quantification of axial curvature defects for all genes at 2 and 5 dpf. (G) Quantification of hemorrhage defects for all genes at 2 and 5 dpf.
Figure 1.
Quantification of phenotypes and gRNA efficacy. (A) Amplicons for each gRNA target were generated by PCR and then TOPO-cloned to generate a library of targeted genomic DNA sequences. (B) Individual clones were amplified by colony PCR and sent for sequencing. (C) Sequences were aligned to wt to determine the incidence of indel mutations. (D) For most targets, we are unable to isolate wt DNA by cloning, indicating a very high degree of mutational efficiency (with the exception of emilin1b gRNA 2, which appears to be non-functional). (E) Quantification of cardiomegaly phenotypes for all genes at 2 and 5 dpf. (F) Quantification of axial curvature defects for all genes at 2 and 5 dpf. (G) Quantification of hemorrhage defects for all genes at 2 and 5 dpf.
Figure 2.
Pedigrees of EMILIN1 (Panel A) and MIB1 (Panel B) families with thoracic aortic disease tested as part of routine clinical genetic testing at the Yale Aortic Institute. For testing EMILIN1 variants in the zebrafish model, missense variants identified in TAAFam02 and TAAFam03 were used (p.E170G and p.T904S, respectively). For testing MIB1 variants in the zebrafish model, nonsense variants identified in TAAFam05, TAAFam06, TAAFam07 were used (p.R906X seen both in TAAFam05 and TAAFam06 and p. G596X for TAAFam07). The index patients (labeled with an arrow) underwent exome sequencing, while other family members underwent single-site Sanger sequencing.
Figure 2.
Pedigrees of EMILIN1 (Panel A) and MIB1 (Panel B) families with thoracic aortic disease tested as part of routine clinical genetic testing at the Yale Aortic Institute. For testing EMILIN1 variants in the zebrafish model, missense variants identified in TAAFam02 and TAAFam03 were used (p.E170G and p.T904S, respectively). For testing MIB1 variants in the zebrafish model, nonsense variants identified in TAAFam05, TAAFam06, TAAFam07 were used (p.R906X seen both in TAAFam05 and TAAFam06 and p. G596X for TAAFam07). The index patients (labeled with an arrow) underwent exome sequencing, while other family members underwent single-site Sanger sequencing.
Figure 3.
Knocking out Ehlers-Danlos and Marfan Syndrome-related genes generates vascular phenotypes. (A) Brightfield macroscopic image of representative series of uninjected larvae at 3 dpf. Scale bar: 1 mm. (B) Similar image of fbn1 knockout fish. Filled arrowhead indicates cardiomegaly. (C) Similar image of col1a2 knockout fish. (D) Similar image of col5a1 knockout fish. (E) Similar image of col5a2a/b knockout fish. (F) Closeup brightfield image of a 6 dpf col5a1 knockout fish with visible aortic hemorrhage (filled arrowhead, scale bar: 500 µm). (G) Confocal micrograph of trunk vasculature of a 4 dpf uninjected Tg(kdr:gfp) fish. DA: dorsal aorta, ISV: intersegmental vessel. Scale bar: 100 µm. (H) Similar micrograph of a col5a1 knockout fish. Empty arrowheads indicate b: excessively branching segment and m: missing segments. (I) Quantification of DA diameter. Diameter is not significantly changed across conditions (t-test, p = 0.998). (J) Quantification of ISV diameter. Diameter is not significantly changed across conditions (t-test, p = 0.342).
Figure 3.
Knocking out Ehlers-Danlos and Marfan Syndrome-related genes generates vascular phenotypes. (A) Brightfield macroscopic image of representative series of uninjected larvae at 3 dpf. Scale bar: 1 mm. (B) Similar image of fbn1 knockout fish. Filled arrowhead indicates cardiomegaly. (C) Similar image of col1a2 knockout fish. (D) Similar image of col5a1 knockout fish. (E) Similar image of col5a2a/b knockout fish. (F) Closeup brightfield image of a 6 dpf col5a1 knockout fish with visible aortic hemorrhage (filled arrowhead, scale bar: 500 µm). (G) Confocal micrograph of trunk vasculature of a 4 dpf uninjected Tg(kdr:gfp) fish. DA: dorsal aorta, ISV: intersegmental vessel. Scale bar: 100 µm. (H) Similar micrograph of a col5a1 knockout fish. Empty arrowheads indicate b: excessively branching segment and m: missing segments. (I) Quantification of DA diameter. Diameter is not significantly changed across conditions (t-test, p = 0.998). (J) Quantification of ISV diameter. Diameter is not significantly changed across conditions (t-test, p = 0.342).
![Genes 13 00123 g003]()
Figure 4.
Knocking out mib1 generates a hemorrhagic phenotype. (A) Genomic region of chromosome 2 containing mib1 gene. Two patient mutations G596 and R906 are indicated. gRNAs (magenta) were designed to target both regions. (B) T7 endonuclease activity against gRNA target amplicons. Uninjected DNA pool (−) produces one fragmentation pattern, mib1-injected DNA pool (+) produces extra bands (filled arrowheads) caused by heteroduplex DNA formation due to editing and subsequent digestion by T7 endonuclease. (C) Brightfield macroscopic image of representative series of uninjected larvae at 3 dpf. Scale bar: 1 mm. (D) Similar image of mib1 knockout fish. Note severe body curvature defects. (E) Uninjected 54 hpf fish, dorsal view. Scale bar: 250 µm. (F) Similar mib1 knockout fish exhibiting cranial hemorrhage (filled arrowhead). (G) Brightfield image of 50 hpf mib1 knockout fish with aortic hemorrhage (filled arrowhead, scale bar: 500 µm). (H) Quantification of hemorrhagic phenotype. Mib1 knockout fish exhibit significantly more hemorrhagic events (uninj: 1%, mib1: 31%, chi-square test, p < 0.001).
Figure 4.
Knocking out mib1 generates a hemorrhagic phenotype. (A) Genomic region of chromosome 2 containing mib1 gene. Two patient mutations G596 and R906 are indicated. gRNAs (magenta) were designed to target both regions. (B) T7 endonuclease activity against gRNA target amplicons. Uninjected DNA pool (−) produces one fragmentation pattern, mib1-injected DNA pool (+) produces extra bands (filled arrowheads) caused by heteroduplex DNA formation due to editing and subsequent digestion by T7 endonuclease. (C) Brightfield macroscopic image of representative series of uninjected larvae at 3 dpf. Scale bar: 1 mm. (D) Similar image of mib1 knockout fish. Note severe body curvature defects. (E) Uninjected 54 hpf fish, dorsal view. Scale bar: 250 µm. (F) Similar mib1 knockout fish exhibiting cranial hemorrhage (filled arrowhead). (G) Brightfield image of 50 hpf mib1 knockout fish with aortic hemorrhage (filled arrowhead, scale bar: 500 µm). (H) Quantification of hemorrhagic phenotype. Mib1 knockout fish exhibit significantly more hemorrhagic events (uninj: 1%, mib1: 31%, chi-square test, p < 0.001).
![Genes 13 00123 g004]()
Figure 5.
Knocking out emilin1a/b reduces DA and ISV diameter, endothelial cell counts. (A) Genomic region of chromosomes 20 and 17 containing emilin1a and b, respectively. Patient mutations E170G and T904S are indicated and their corresponding gRNAs are overlaid (magenta). (B) T7 endonuclease activity against gRNA target amplicons for emilin1a. Supernumerary bands are generated for both injected gRNAs (filled arrowheads). (C) Similar T7 assay as (B), but applied to emilin1b target amplicons. (D) Brightfield macroscopic image of a representative series of uninjected 3 dpf larvae. Scale bar: 1 mm. (E) Similar image of emilin1a/b fish. Note that fish are generally normal in appearance. (F) Confocal micrograph of uninjected 3 dpf Tg(kdr:gfp) trunk vasculature. DA: dorsal aorta, ISV: intersegmental vessel, scale bar: 100 µm. (G) Similar micrograph taken of an emilin1a/b knockout fish. DA is traced in blue to emphasize tortuosity. Note the generally lower diameter of vessels, (H) Uninjected 3 dpf Tg(fli1a:nls-gfp); Tg(kdrl:ras-mcherry) trunk vasculature. DA: dorsal aorta, ISV: intersegmental vessel. Endothelial cell nuclei are labelled with GFP, making quantification possible. (I) Similar micrograph taken from an emilin1a/b knockout fish. These parameters are qualified in the following five panels: (J) DA diameter, (K) ISV diameter, (L) DA endothelial cell count, (M) ISV endothelial cell count, (N) DA tortuosity. With the exception of (N) all parameters are significant as indicated (t-test).
Figure 5.
Knocking out emilin1a/b reduces DA and ISV diameter, endothelial cell counts. (A) Genomic region of chromosomes 20 and 17 containing emilin1a and b, respectively. Patient mutations E170G and T904S are indicated and their corresponding gRNAs are overlaid (magenta). (B) T7 endonuclease activity against gRNA target amplicons for emilin1a. Supernumerary bands are generated for both injected gRNAs (filled arrowheads). (C) Similar T7 assay as (B), but applied to emilin1b target amplicons. (D) Brightfield macroscopic image of a representative series of uninjected 3 dpf larvae. Scale bar: 1 mm. (E) Similar image of emilin1a/b fish. Note that fish are generally normal in appearance. (F) Confocal micrograph of uninjected 3 dpf Tg(kdr:gfp) trunk vasculature. DA: dorsal aorta, ISV: intersegmental vessel, scale bar: 100 µm. (G) Similar micrograph taken of an emilin1a/b knockout fish. DA is traced in blue to emphasize tortuosity. Note the generally lower diameter of vessels, (H) Uninjected 3 dpf Tg(fli1a:nls-gfp); Tg(kdrl:ras-mcherry) trunk vasculature. DA: dorsal aorta, ISV: intersegmental vessel. Endothelial cell nuclei are labelled with GFP, making quantification possible. (I) Similar micrograph taken from an emilin1a/b knockout fish. These parameters are qualified in the following five panels: (J) DA diameter, (K) ISV diameter, (L) DA endothelial cell count, (M) ISV endothelial cell count, (N) DA tortuosity. With the exception of (N) all parameters are significant as indicated (t-test).
![Genes 13 00123 g005]()
Table 1.
scRNAs and primers used to perform T7 assays. For each gene, two target-specific scRNAs were designed. Flanking primers generating amplicons between 300–600 bp were also generated to assess whether indel mutations were being generated at the desired locations.
Table 1.
scRNAs and primers used to perform T7 assays. For each gene, two target-specific scRNAs were designed. Flanking primers generating amplicons between 300–600 bp were also generated to assess whether indel mutations were being generated at the desired locations.
| Target Gene | scRNA Sequence | | Forward Primer | | Reverse Primer |
|---|
| col1a2 | CCTGGCCCCCCTGGTCTTGG | 5′ | CCAGTTGTTTGATTATATTGTTCTG | 5′ | GTCTCAAATATAACAACAAACACTC |
| col1a2 | CAGCACCGCGAGCACCAGCT | 5′ | CTACTTATTTGAACTGTTCACTTTG | 5′ | TAATCTTCTAGCCACATTATTTCAC |
| col5a1 | CCGAAACGGAACTTCTGGAT | 5′ | GATATGAAACCTATTATGACGAGTC | 5′ | TAGATAACTAAACTCACTGAGGTAG |
| col5a1 | AGCATCGGTGTAATCCGTGA | 5′ | GAGAAGGAGATTATACTGTAGGAG | 5′ | CTGTTATAAATGAACAAGTCATTCC |
| col5a2a | TGCCAGGGCGATGCAGCTGA | 5′ | GATGAGTTGAGCTGTACAGAG | 5′ | AAGAAGAAACATAACAGACAAAAAC |
| col5a2a | AGGCACTAATGCCCGGTGTT | 5′ | GCAGATCCAATAAAGTGATTATTTC | 5′ | TGTGGACAAATTATTCGTTTTTATC |
| col5a2b | CGAGCAATTCTTGATCCCGG | 5′ | GCAATTGAAACAAACACAATATAAC | 5′ | GTATTTAACGTGACGTTAAGTTAAC |
| col5a2b | CCTATAGGACCAGGCTCACC | 5′ | TTCTTATTTAGATTGTGGGAATACC | 5′ | GCACATTTTATTACTCTAATCACTG |
| emilin1a | GCAAGACCTACTGGTGAAGA | 5′ | TGATTAATTGTTATGTTGATGCATG | 5′ | AAAGTGTTACCAAATAAACAAACTC |
| emilin1a | AAGGCCTACAATCCAAAAAC | 5′ | CAGTAATCTAACTAATACAACTCCC | 5′ | ATAACATTGTTAGCATTTAACACTG |
| emilin1b | CAGGCAGACCTGGGAATGGT | 5′ | TTTGAAGTGAAAGTTCTGTTTAATG | 5′ | TGCTAAGATGATATATTCAGTGTTG |
| emilin1b | AACTTTTACAATTCACGAAC | 5′ | TTTGTATGAGAGTCAGTGATATTTC | 5′ | CTAAAGAAGTACTTTCCTTCTACTG |
| fbn1 | ATACCGCTGCCTCTGCAAAG | 5′ | CTCATAACTGTGATCATCATGC | 5′ | CTCTCACACAGATTCACATTATC |
| fbn1 | GAGCTCAATCCCACTGGAGT | 5′ | ATACCCATGTCATTATACAGATTTG | 5′ | CCAAATTATCCAGCTTATGTTTTAC |
| mib1 | GTCACAATCACCAACAACAA | 5′ | TCTCTTTCTTGATTTTTGAAATTGG | 5′ | TACAGAAGGATTGAATAATTCTGAC |
| mib1 | TGTGCAGTGTCGGGCCGTGG | 5′ | CAAAATTAAATGTCATACAGTGAGG | 5′ | AAATATTGAAGATGAGAGACATGAG |
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}