
Wild-Type KRAS Allele Effects on Druggable Targets in KRAS Mutant Lung Adenocarcinomas

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. NSCLC Cell Lines and Cultures
2.2. Cell Viability Assay
2.3. Cell Lysate Preparation for Signaling Network Analysis
2.4. NSCLC Tissue Collection
2.5. Laser Capture Microdissection
2.6. DNA Extraction and PCR Amplification
2.7. Reverse-Phase Protein Microarray
2.8. Clonogenic Assay
2.9. Statistical Analysis
3. Results
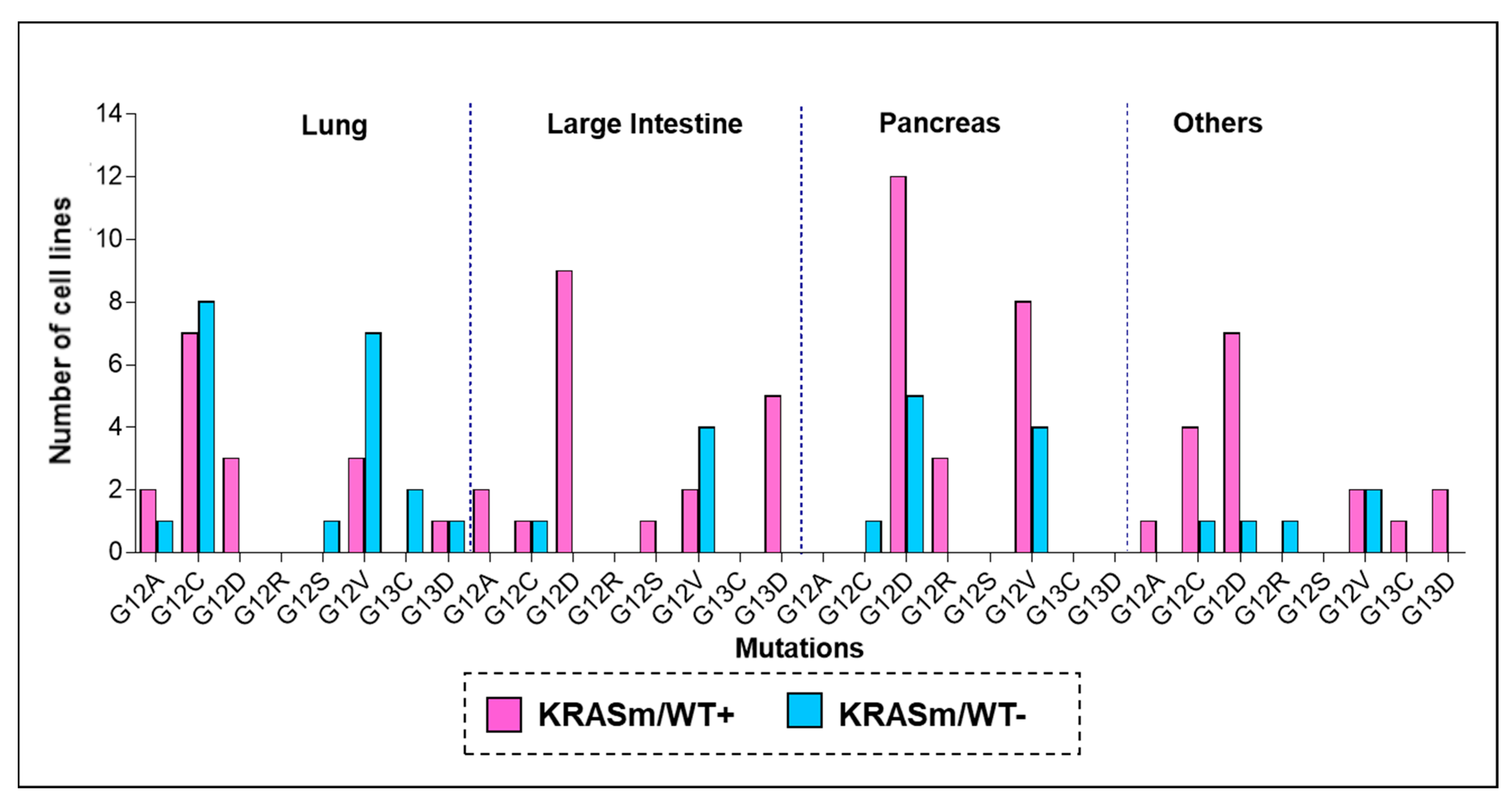
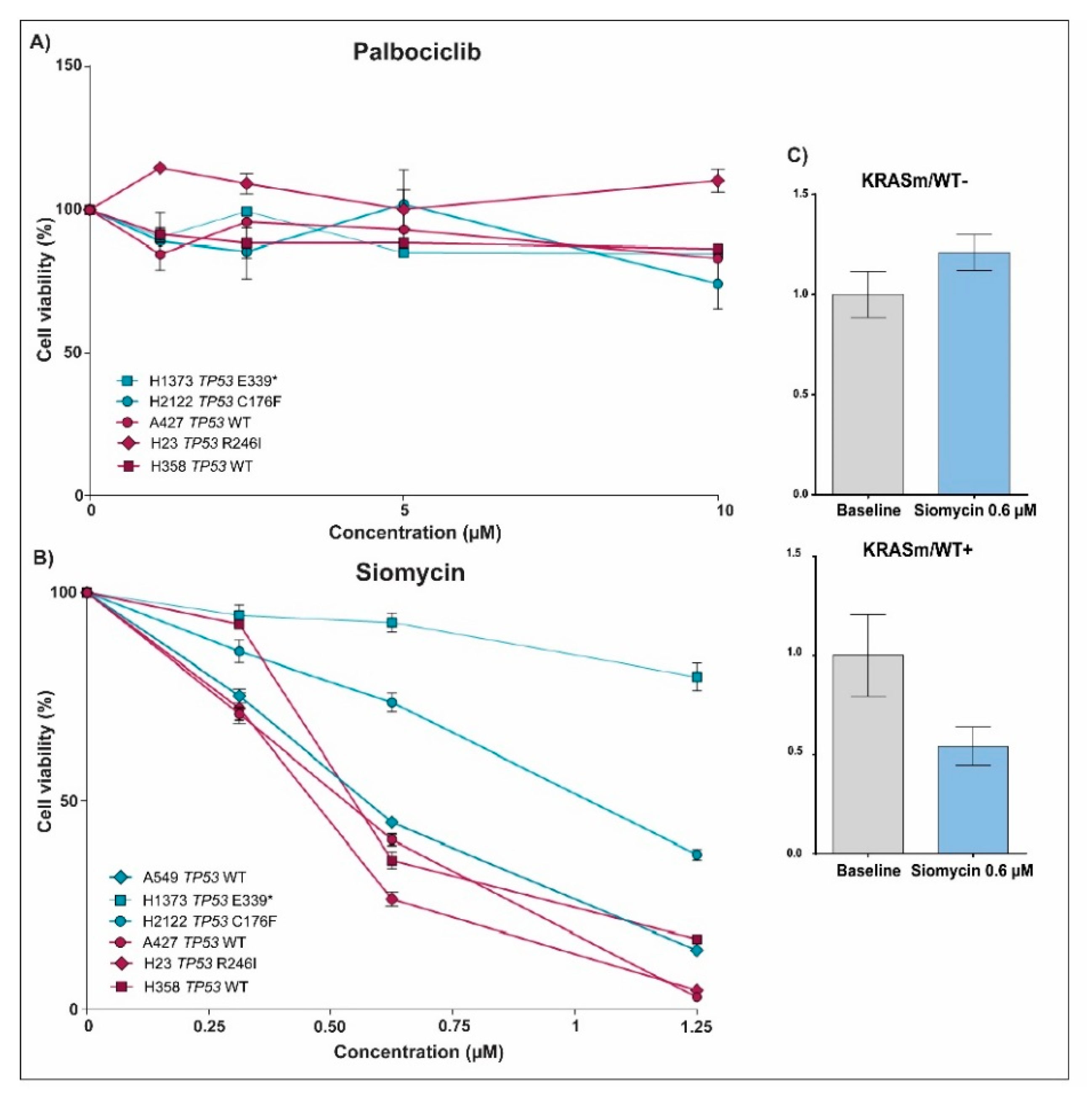
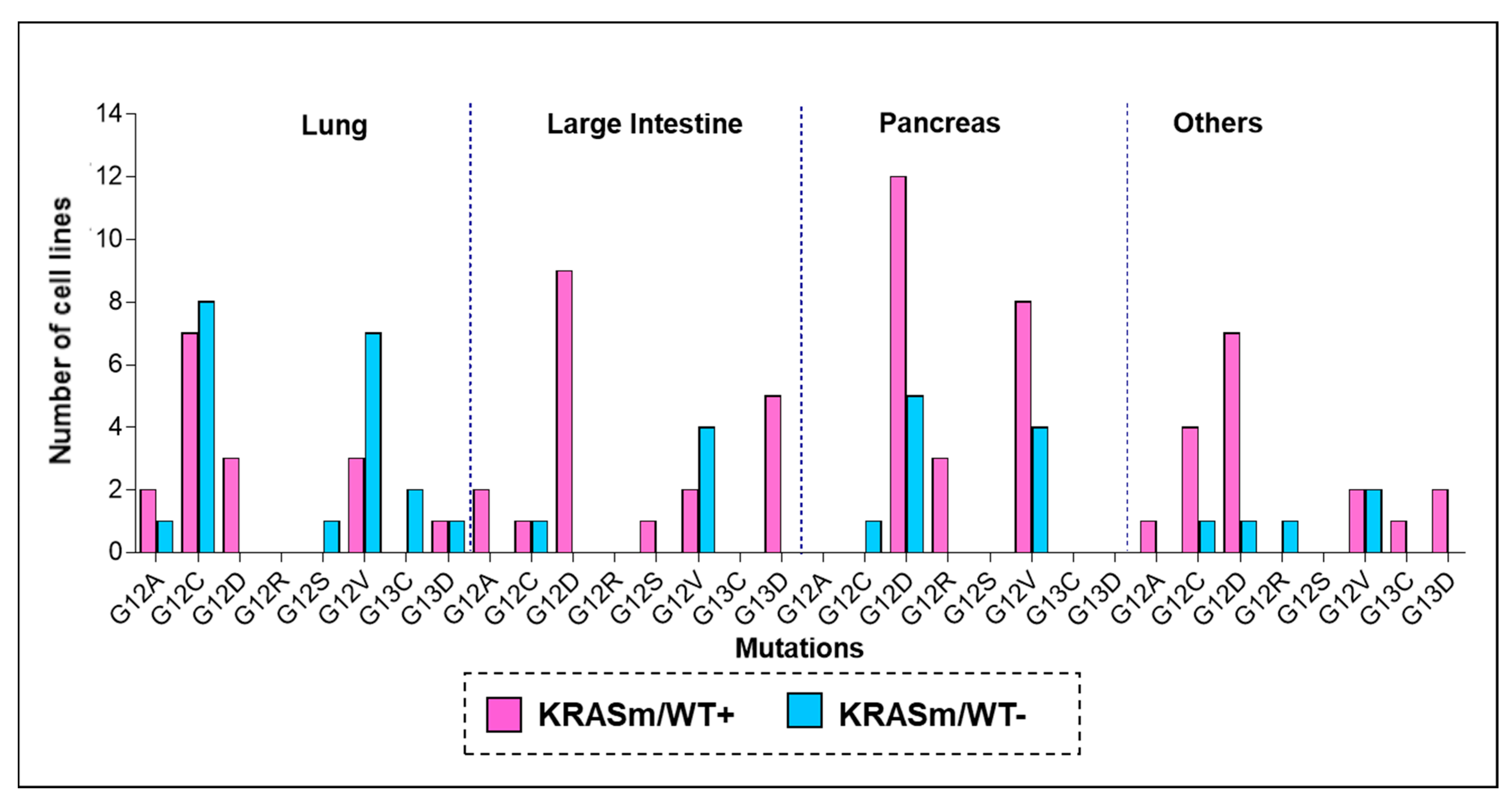
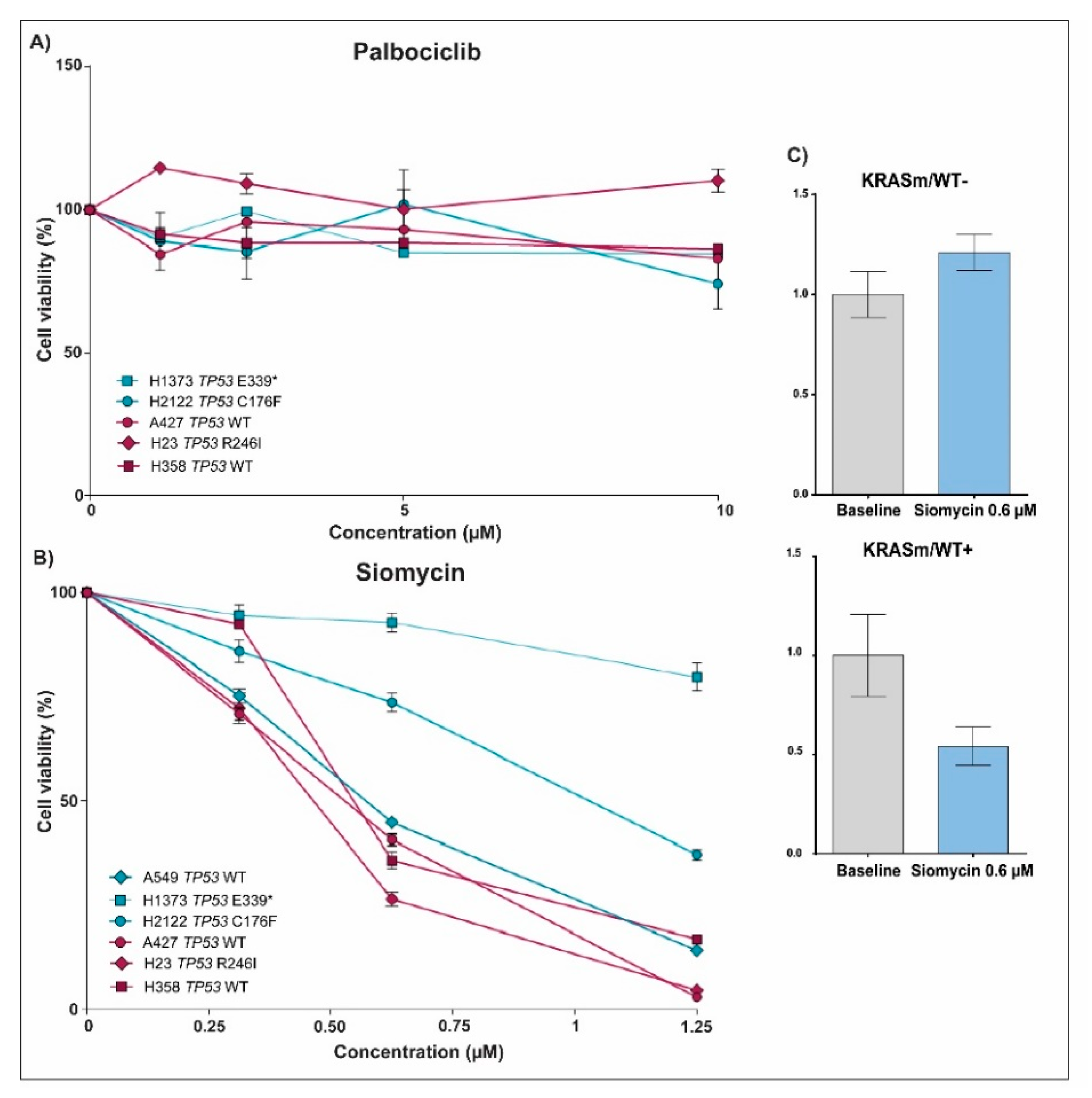
3.1. Response to Treatment in KRAS Mutant NSCLC Is Affected by the Presence of the WT KRAS Allele
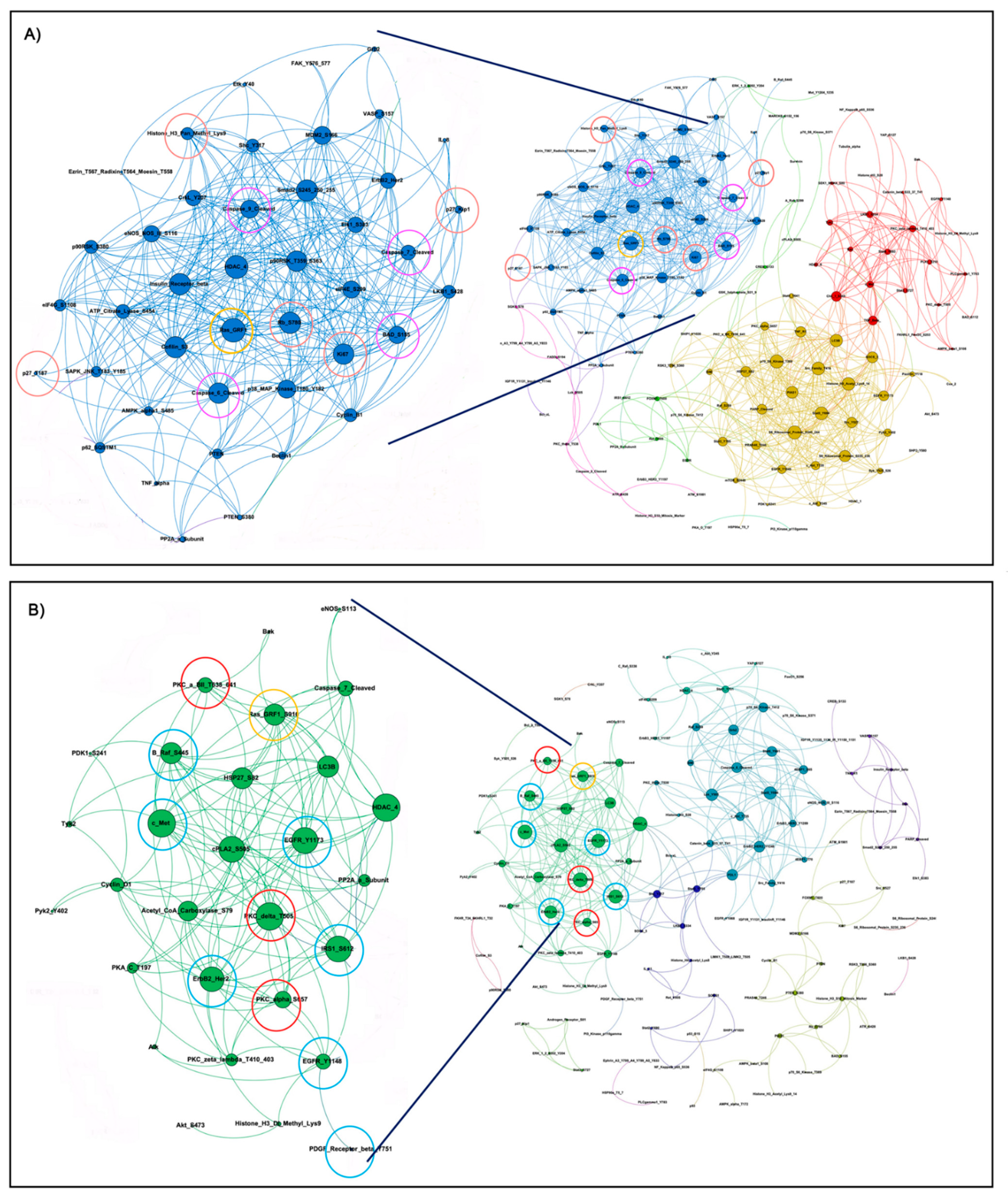
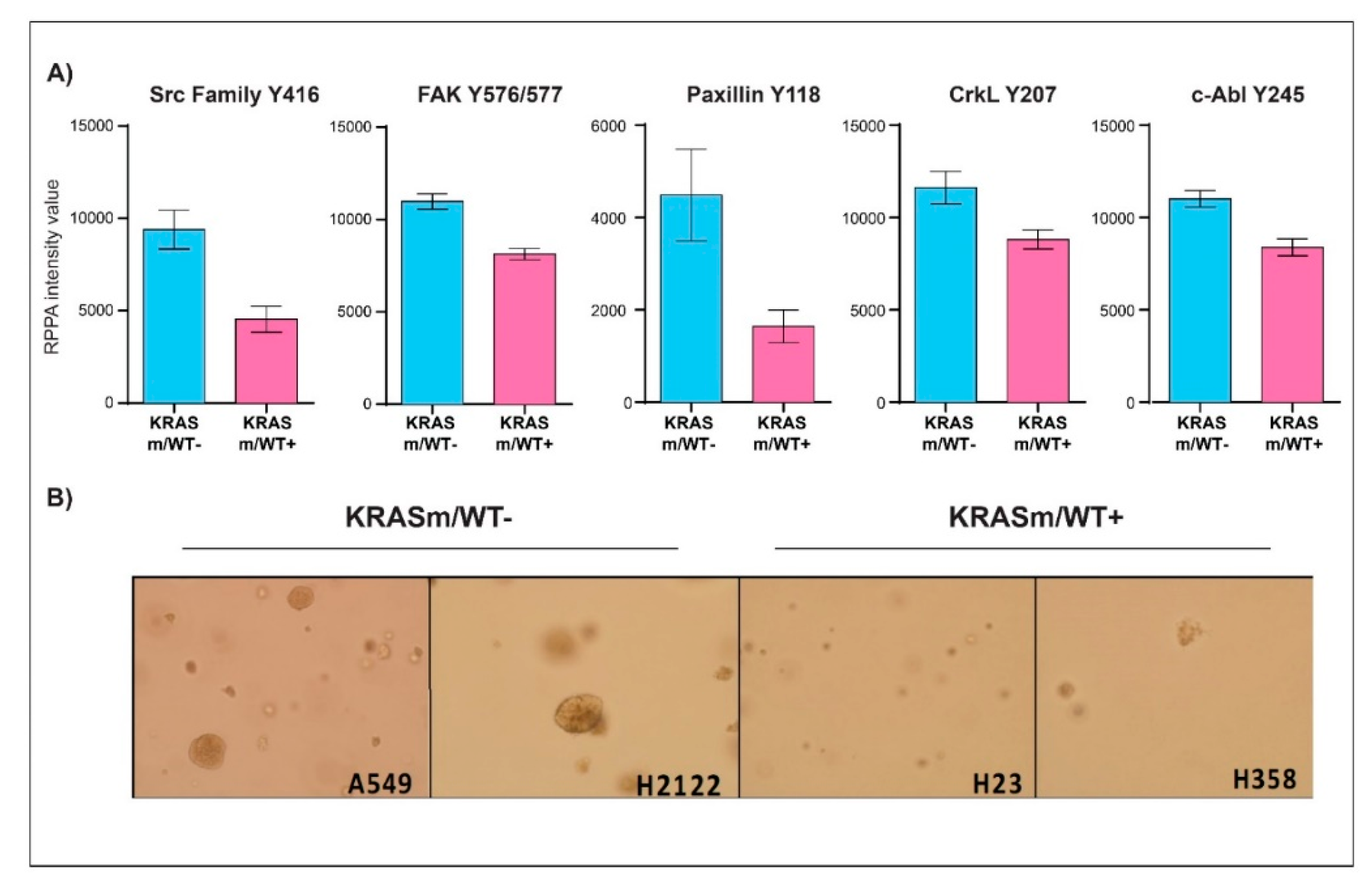
3.2. KRAS WT Alleles Drive Specific Signaling Alterations in Lung Adenocarcinomas Harboring KRAS Oncogenic Mutations
3.3. KRAS Mutant Lung Adenocarcinoma Cells Retaining the WT KRAS Allele Have Aberrant Activation of Cell Cycle Regulators and DNA Repair Mechanisms
3.4. FoxM1 Activation Is Increased in Surgical Specimens of NSCLC Patients Harboring KRASm/WT+ Lesions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wang, Y.; Li, X. Targeting the Untargetable KRAS in Cancer Therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef] [PubMed]
- Crul, M.; de Klerk, G.J.; Swart, M.; van’t Veer, L.J.; de Jong, D.; Boerrigter, L.; Palmer, P.A.; Bol, C.J.; Tan, H.; de Gast, G.C.; et al. Phase I Clinical and Pharmacologic Study of Chronic Oral Administration of the Farnesyl Protein Transferase Inhibitor R115777 in Advanced Cancer. JCO 2002, 20, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- McDermott, R.S.; Meropol, N.J.; Weiner, L.M.; Yeslow, G.; Hudes, G.R. Phase I Clinical and Pharmacokinetic Study of the Farnesyl Transferase Inhibitor BMS-214662 Administered Intravenously for Five Consecutive Days in Patients with Advanced Malignancies. JCO 2004, 22, 2033. [Google Scholar] [CrossRef]
- Zujewski, J.; Horak, I.D.; Bol, C.J.; Woestenborghs, R.; Bowden, C.; End, D.W.; Piotrovsky, V.K.; Chiao, J.; Belly, R.T.; Todd, A.; et al. Phase I and Pharmacokinetic Study of Farnesyl Protein Transferase Inhibitor R115777 in Advanced Cancer. JCO 2000, 18, 927. [Google Scholar] [CrossRef]
- Appels, N.M.G.M.; Beijnen, J.H.; Schellens, J.H.M. Development of Farnesyl Transferase Inhibitors: A Review. Oncologist 2005, 10, 565–578. [Google Scholar] [CrossRef] [Green Version]
- Kohler, J.; Catalano, M.; Ambrogio, C. Back to the Bench? MEK and ERK Inhibitors for the Treatment of KRAS Mutant Lung Adenocarcinoma. CMC 2018, 25, 558–574. [Google Scholar] [CrossRef]
- Jänne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS -Mutant Advanced Non–Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017, 317, 1844. [Google Scholar] [CrossRef] [Green Version]
- Blumenschein, G.R.; Smit, E.F.; Planchard, D.; Kim, D.-W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.-J.; Hanna, N.H.; et al. A Randomized Phase II Study of the MEK1/MEK2 Inhibitor Trametinib (GSK1120212) Compared with Docetaxel in KRAS-Mutant Advanced Non-Small-Cell Lung Cancer (NSCLC). Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kuo, J.; Sacher, A.G.; Barlesi, F.; Besse, B.; Kuboki, Y.; Dy, G.K.; Dembla, V.; Krauss, J.C.; Burns, T.F.; et al. CodeBreak 100: Phase I Study of AMG 510, a Novel KRAS G12C Inhibitor, in Patients (Pts) with Advanced Solid Tumors Other than Non-Small Cell Lung Cancer (NSCLC) and Colorectal Cancer (CRC). JCO 2020, 38, 3511. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Govindan, R.; Dy, G.K.; Shapiro, G.; Bauml, J.; Schuler, M.H.; Addeo, A.; Kato, T.; Besse, B.; et al. Overall Survival and Exploratory Subgroup Analyses from the Phase 2 CodeBreaK 100 Trial Evaluating Sotorasib in Pretreated KRAS p.G12C Mutated Non-Small Cell Lung Cancer. JCO 2021, 39, 9003. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Rybkin, I.I.; Spira, A.I.; Riely, G.J.; Papadopoulos, K.P.; Sabari, J.K.; Johnson, M.L.; Heist, R.S.; Bazhenova, L.; Barve, M.; et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/ Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur. J. Cancer 2020, 138, S1–S2. [Google Scholar] [CrossRef]
- Doherty, G.J.; Kerr, E.M.; Martins, C.P. KRAS Allelic Imbalance: Strengths and Weaknesses in Numbers. Trends Mol. Med. 2017, 23, 377–378. [Google Scholar] [CrossRef] [Green Version]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L.; et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 2017, 168, 817–829.e15. [Google Scholar] [CrossRef] [Green Version]
- Baldelli, E.; Subramanian, M.; Alsubaie, A.M.; Oldaker, G.; Emelianenko, M.; El Gazzah, E.; Baglivo, S.; Hodge, K.A.; Bianconi, F.; Ludovini, V.; et al. Heterogeneous Off-Target Effects of Ultra-Low Dose Dimethyl Sulfoxide (DMSO) on Targetable Signaling Events in Lung Cancer In Vitro Models. Int. J. Mol. Sci. 2021, 22, 2819. [Google Scholar] [CrossRef]
- Ludovini, V.; Bianconi, F.; Pistola, L.; Chiari, R.; Minotti, V.; Colella, R.; Giuffrida, D.; Tofanetti, F.R.; Siggillino, A.; Flacco, A.; et al. Phosphoinositide-3-Kinase Catalytic α and KRAS Mutations Are Important Predictors of Resistance to Therapy with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients with Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Sereni, M.I.; Baldelli, E.; Gambara, G.; Ravaggi, A.; Hodge, K.A.; Alberts, D.S.; Guillen-Rodriguez, J.M.; Dong, T.; Memo, M.; Odicino, F.; et al. Kinase-Driven Metabolic Signalling as a Predictor of Response to Carboplatin-Paclitaxel Adjuvant Treatment in Advanced Ovarian Cancers. Br. J. Cancer 2017, 117, 494–502. [Google Scholar] [CrossRef]
- Baldelli, E.; Calvert, V.; Hodge, A.; VanMeter, A.; Petricoin, E.F.; Pierobon, M. Reverse Phase Protein Microarrays. Methods Mol. Biol. 2017, 1606, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Signore, M.; Manganelli, V.; Hodge, A. Antibody Validation by Western Blotting. Methods Mol. Biol. 2017, 1606, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The Soft Agar Colony Formation Assay. J. Vis. Exp. 2014, 92, 51998. [Google Scholar] [CrossRef] [Green Version]
- Ambrogio, C.; Köhler, J.; Zhou, Z.-W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.; Hensing, T.; Malik, R.; Salgia, R. Prognostic and Predictive Value in KRAS in Non–Small-Cell Lung Cancer: A Review. JAMA Oncol. 2016, 2, 805. [Google Scholar] [CrossRef]
- Soria, J.-C.; Fülöp, A.; Maciel, C.; Fischer, J.R.; Girotto, G.; Lago, S.; Smit, E.; Ostoros, G.; Eberhardt, W.E.E.; Lishkovska, P.; et al. SELECT-2: A Phase II, Double-Blind, Randomized, Placebo-Controlled Study to Assess the Efficacy of Selumetinib plus Docetaxel as a Second-Line Treatment of Patients with Advanced or Metastatic Non-Small-Cell Lung Cancer. Ann. Oncol. 2017, 28, 3028–3036. [Google Scholar] [CrossRef]
- Koo, C.-Y.; Muir, K.W.; Lam, E.W.-F. FOXM1: From Cancer Initiation to Progression and Treatment. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1819, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-M.; Ackerson, T.; Ramakrishna, S.; Tretiakova, M.; Wang, I.-C.; Kalin, T.V.; Major, M.L.; Gusarova, G.A.; Yoder, H.M.; Costa, R.H.; et al. The Forkhead Box M1 Transcription Factor Stimulates the Proliferation of Tumor Cells during Development of Lung Cancer. Cancer Res. 2006, 66, 2153–2161. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Wang, I.; Yoder, H.M.; Davidson, N.O.; Costa, R.H. The Forkhead Box M1 Transcription Factor Contributes to the Development and Growth of Mouse Colorectal Cancer. Gastroenterology 2007, 132, 1420–1431. [Google Scholar] [CrossRef]
- Song, X.; Fiati Kenston, S.S.; Zhao, J.; Yang, D.; Gu, Y. Roles of FoxM1 in Cell Regulation and Breast Cancer Targeting Therapy. Med. Oncol. 2017, 34, 41. [Google Scholar] [CrossRef]
- Zhao, F.; Lam, E.W.-F. Role of the Forkhead Transcription Factor FOXO-FOXM1 Axis in Cancer and Drug Resistance. Front. Med. 2012, 6, 376–380. [Google Scholar] [CrossRef]
- Liao, G.-B.; Li, X.-Z.; Zeng, S.; Liu, C.; Yang, S.-M.; Yang, L.; Hu, C.-J.; Bai, J.-Y. Regulation of the Master Regulator FOXM1 in Cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.Y.M.; Tong, T.H.K.; Leung, W.Y.; Yao, K.-M. Raf/MEK/MAPK Signaling Stimulates the Nuclear Translocation and Transactivating Activity of FOXM1. In Transcription Factors; Higgins, P.J., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volme 647, pp. 113–123. ISBN 978-1-60761-737-2. [Google Scholar]
- Radhakrishnan, S.K.; Bhat, U.G.; Hughes, D.E.; Wang, I.-C.; Costa, R.H.; Gartel, A.L. Identification of a Chemical Inhibitor of the Oncogenic Transcription Factor Forkhead Box M1. Cancer Res. 2006, 66, 9731–9735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovly, C.M.; Salama, A.K.S.; Salgia, R. Tumor Heterogeneity and Therapeutic Resistance. Am. Soc. Clin. Oncol. Educ. Book 2016, 36, e585–e593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Späth, S.S.; Marjani, S.L.; Zhang, W.; Pan, X. Characterization of Cancer Genomic Heterogeneity by Next-Generation Sequencing Advances Precision Medicine in Cancer Treatment. Precis. Clin. Med. 2018, 1, 29–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gao, G.F.; Minna, J.D.; Williams, N.S.; Westover, K.D. Loss of Wild Type KRAS in KRAS Lung Adenocarcinoma Is Associated with Cancer Mortality and Confers Sensitivity to FASN Inhibitors. Lung Cancer 2021, 153, 73–80. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldelli, E.; El Gazzah, E.; Moran, J.C.; Hodge, K.A.; Manojlovic, Z.; Bassiouni, R.; Carpten, J.D.; Ludovini, V.; Baglivo, S.; Crinò, L.; et al. Wild-Type KRAS Allele Effects on Druggable Targets in KRAS Mutant Lung Adenocarcinomas. Genes 2021, 12, 1402. https://doi.org/10.3390/genes12091402
Baldelli E, El Gazzah E, Moran JC, Hodge KA, Manojlovic Z, Bassiouni R, Carpten JD, Ludovini V, Baglivo S, Crinò L, et al. Wild-Type KRAS Allele Effects on Druggable Targets in KRAS Mutant Lung Adenocarcinomas. Genes. 2021; 12(9):1402. https://doi.org/10.3390/genes12091402
Chicago/Turabian StyleBaldelli, Elisa, Emna El Gazzah, John Conor Moran, Kimberley A. Hodge, Zarko Manojlovic, Rania Bassiouni, John D. Carpten, Vienna Ludovini, Sara Baglivo, Lucio Crinò, and et al. 2021. "Wild-Type KRAS Allele Effects on Druggable Targets in KRAS Mutant Lung Adenocarcinomas" Genes 12, no. 9: 1402. https://doi.org/10.3390/genes12091402
APA StyleBaldelli, E., El Gazzah, E., Moran, J. C., Hodge, K. A., Manojlovic, Z., Bassiouni, R., Carpten, J. D., Ludovini, V., Baglivo, S., Crinò, L., Bianconi, F., Dong, T., Loffredo, J., Petricoin, E. F., & Pierobon, M. (2021). Wild-Type KRAS Allele Effects on Druggable Targets in KRAS Mutant Lung Adenocarcinomas. Genes, 12(9), 1402. https://doi.org/10.3390/genes12091402