
Alternative Splicing Role in New Therapies of Spinal Muscular Atrophy



Abstract
1. Introduction
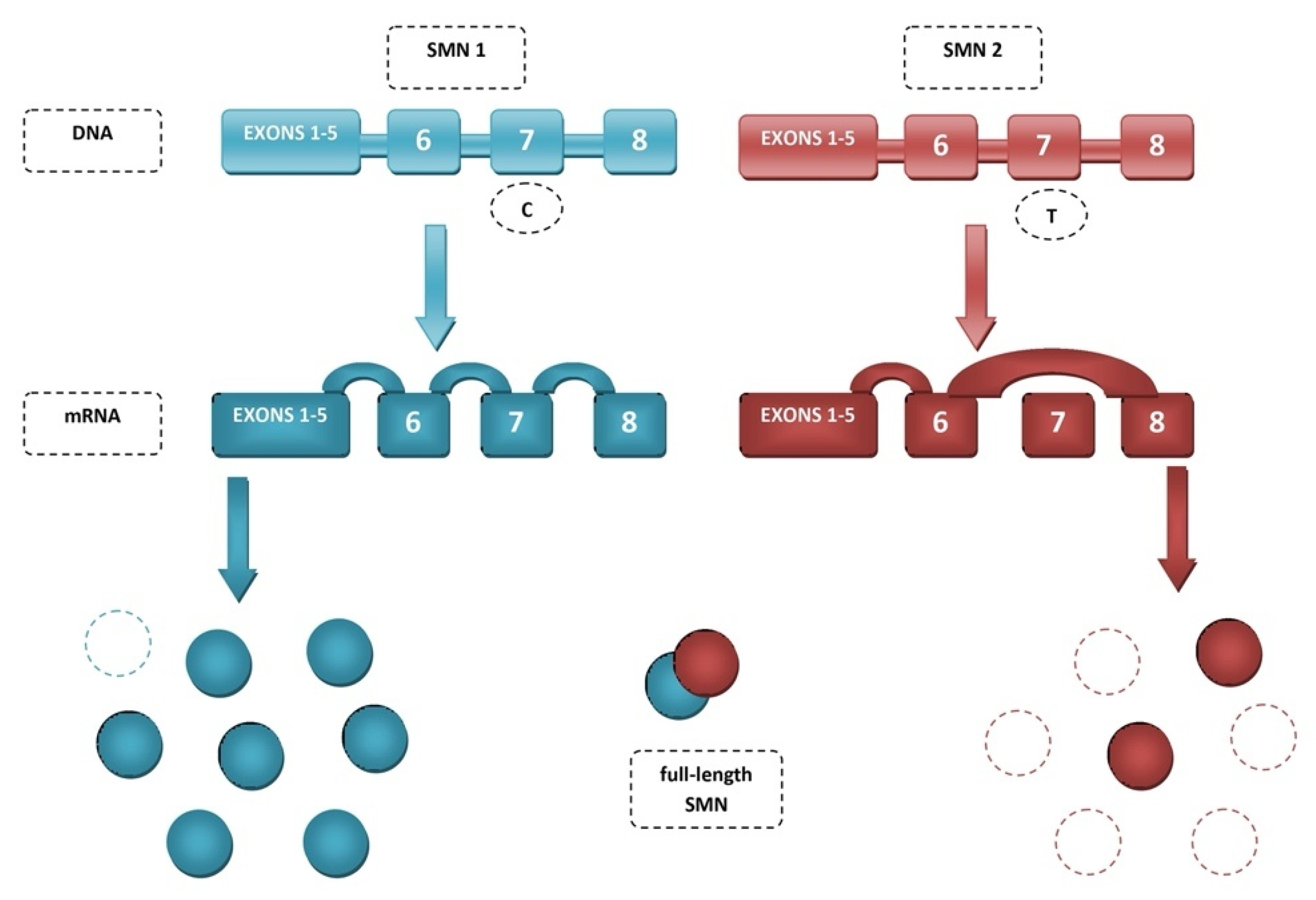
2. Spinal Muscular Atrophy (SMA)
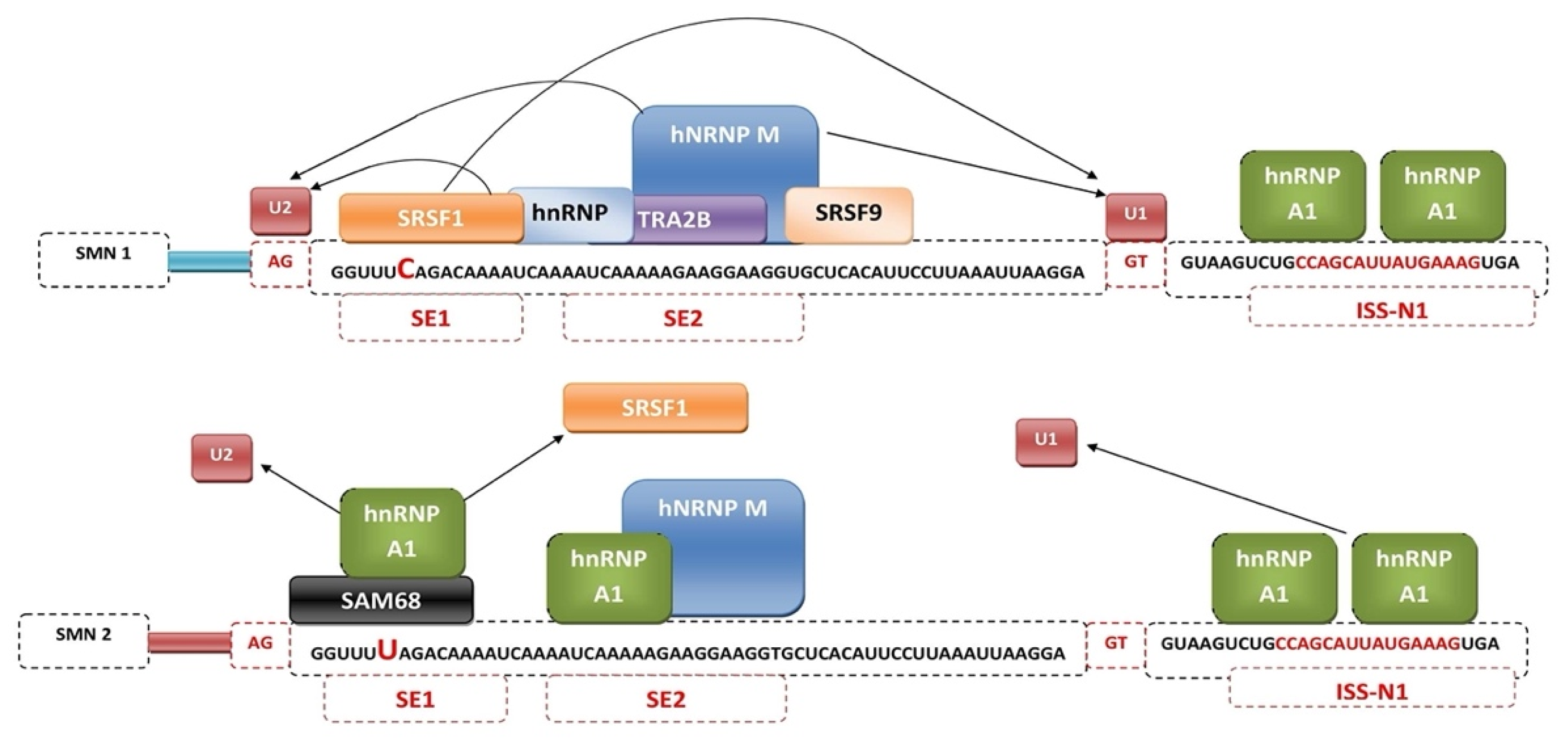
3. Mechanisms of SMN2 Splicing Regulation Targeted by Therapeutics
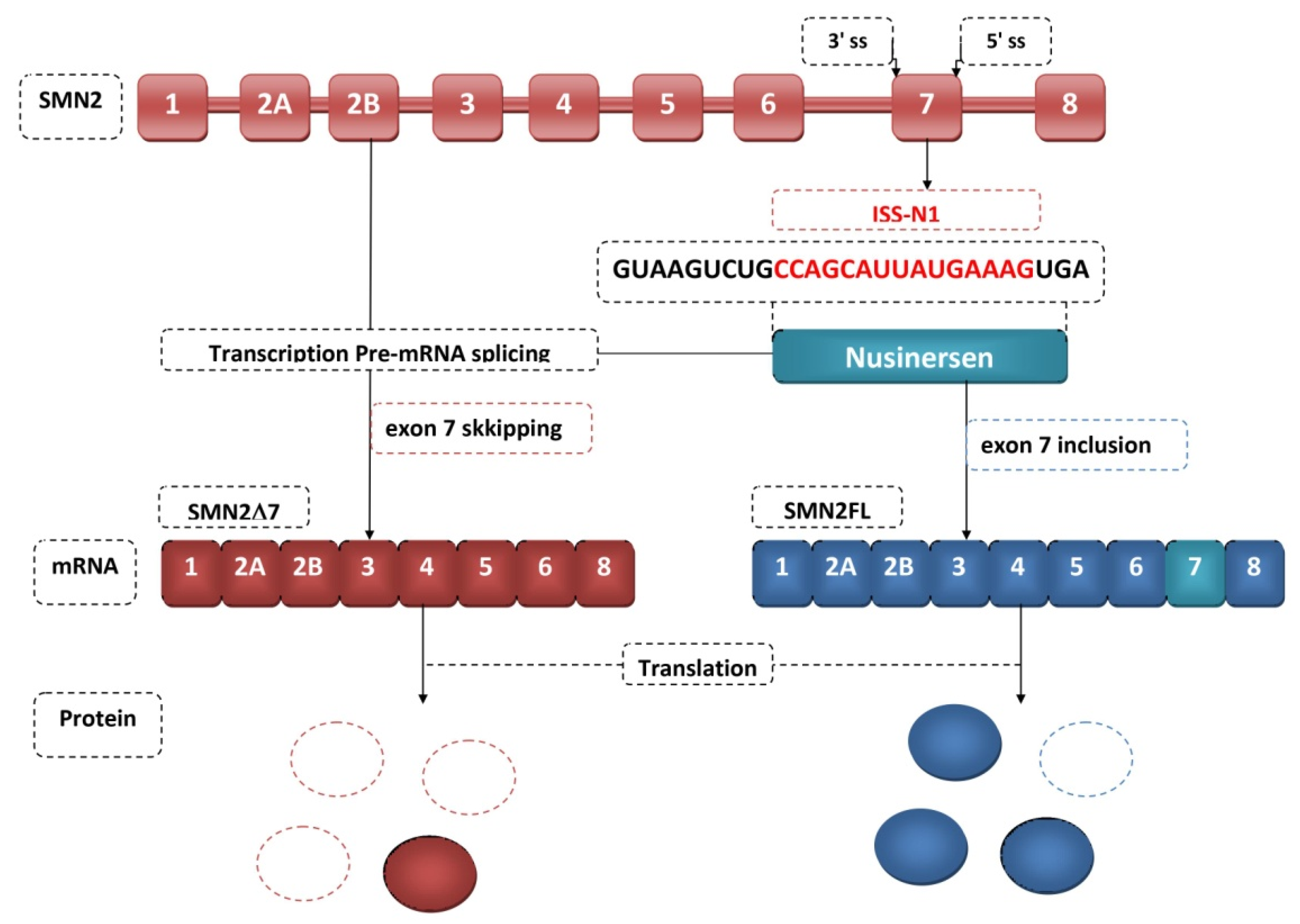
4. Antisense Oligonucleotides
5. Small Molecules
6. Future Prospects
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faustino, N.A.; Cooper, T.A. Pre-MRNA Splicing and Human Disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke Off. J. Int. Stroke Soc. 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z. Systematical Identification of Splicing Regulatory Cis-Elements and Cognate Trans-Factors. Methods 2014, 65, 350–358. [Google Scholar] [CrossRef]
- Black, A.J.; Gamarra, J.R.; Giudice, J. More than a Messenger: Alternative Splicing as a Therapeutic Target. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194395. [Google Scholar] [CrossRef]
- Montes, M.; Sanford, B.L.; Comiskey, D.F.; Chandler, D.S. RNA Splicing and Disease: Animal Models to Therapies. Trends Genet. TIG 2019, 35, 68–87. [Google Scholar] [CrossRef]
- Long, Y.; Sou, W.H.; Yung, K.W.Y.; Liu, H.; Wan, S.W.C.; Li, Q.; Zeng, C.; Law, C.O.K.; Chan, G.H.C.; Lau, T.C.K.; et al. Distinct Mechanisms Govern the Phosphorylation of Different SR Protein Splicing Factors. J. Biol. Chem. 2019, 294, 1312–1327. [Google Scholar] [CrossRef]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and Disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Singh, N.N.; Ottesen, E.W.; Sivanesan, S.; Shishimorova, M.; Singh, R.N. Oxidative Stress Triggers Body-Wide Skipping of Multiple Exons of the Spinal Muscular Atrophy Gene. PLoS ONE 2016, 11, e0154390. [Google Scholar] [CrossRef] [PubMed]
- Deschênes, M.; Chabot, B. The Emerging Role of Alternative Splicing in Senescence and Aging. Aging Cell 2017, 16, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Apicco, D.J.; Zhang, C.; Maziuk, B.; Jiang, L.; Ballance, H.I.; Boudeau, S.; Ung, C.; Li, H.; Wolozin, B. Dysregulation of RNA Splicing in Tauopathies. Cell Rep. 2019, 29, 4377–4388.e4. [Google Scholar] [CrossRef]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted Alternative Splicing for Genes Implicated in Splicing and Ciliogenesis Causes PRPF31 Retinitis Pigmentosa. Nat. Commun. 2018, 9, 4234. [Google Scholar] [CrossRef]
- Perrone, B.; La Cognata, V.; Sprovieri, T.; Ungaro, C.; Conforti, F.L.; Andò, S.; Cavallaro, S. Alternative Splicing of ALS Genes: Misregulation and Potential Therapies. Cell. Mol. Neurobiol. 2020, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Aretz, S.; Uhlhaas, S.; Sun, Y.; Pagenstecher, C.; Mangold, E.; Caspari, R.; Möslein, G.; Schulmann, K.; Propping, P.; Friedl, W. Familial Adenomatous Polyposis: Aberrant Splicing Due to Missense or Silent Mutations in the APC Gene. Hum. Mutat. 2004, 24, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Coulson, R.L.; Powell, W.T.; Yasui, D.H.; Dileep, G.; Resnick, J.; LaSalle, J.M. Prader-Willi Locus Snord116 RNA Processing Requires an Active Endogenous Allele and Neuron-Specific Splicing by Rbfox3/NeuN. Hum. Mol. Genet. 2018, 27, 4051–4060. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, J.; Zhang, W.; Chen, D.; Wang, Y. Aberrant Alternative Splicing in Breast Cancer. J. Mol. Cell Biol. 2019, 11, 920–929. [Google Scholar] [CrossRef]
- Papatsirou, M.; Adamopoulos, P.G.; Artemaki, P.I.; Georganti, V.P.; Scorilas, A.; Vassilacopoulou, D.; Kontos, C.K. Next-Generation Sequencing Reveals Alternative L-DOPA Decarboxylase (DDC) Splice Variants Bearing Novel Exons, in Human Hepatocellular and Lung Cancer Cells. Gene 2021, 768, 145262. [Google Scholar] [CrossRef]
- Chen, T.-H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int. J. Mol. Sci. 2020, 21, 3297. [Google Scholar] [CrossRef]
- Werdnig, G. Two Early Infantile Hereditary Cases of Progressive Muscular Atrophy Simulating Dystrophy, but on a Neural Basis. 1891. Arch. Neurol. 1971, 25, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J. Ueber chronische spinale Muskelatrophie im Kindesalter, auf familiärer Basis. Dtsch. Z. Nervenheilkd. 1893, 3, 427–470. [Google Scholar] [CrossRef]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, Incidence and Carrier Frequency of 5q–Linked Spinal Muscular Atrophy—A Literature Review. Orphanet J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Burr, P.; Reddivari, A.K.R. Spinal Muscle Atrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Crawford, T.O.; Pardo, C.A. The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef]
- Zaldívar, T.; Montejo, Y.; Acevedo, A.M.; Guerra, R.; Vargas, J.; Garofalo, N.; Alvarez, R.; Alvarez, M.A.; Hardiman, O. Evidence of Reduced Frequency of Spinal Muscular Atrophy Type I in the Cuban Population. Neurology 2005, 65, 636–638. [Google Scholar] [CrossRef]
- Jak Częste Jest SMA? Fund SMA. Available online: https://www.fsma.pl/rdzeniowy-zanik-miesni/jak-czeste-jest-sma/ (accessed on 24 May 2021).
- Jędrzejowska, M.; Kostera-Pruszczyk, A. Spinal Muscular Atrophy—New Therapies, New Challenges. Neurol. Neurochir. Pol. 2020, 54, 8–13. [Google Scholar] [CrossRef]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Alías, L.; Bernal, S.; Fuentes-Prior, P.; Barceló, M.J.; Also, E.; Martínez-Hernández, R.; Rodríguez-Alvarez, F.J.; Martín, Y.; Aller, E.; Grau, E.; et al. Mutation Update of Spinal Muscular Atrophy in Spain: Molecular Characterization of 745 Unrelated Patients and Identification of Four Novel Mutations in the SMN1 Gene. Hum. Genet. 2009, 125, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Hahnen, E.; Morgan, K.; DiDonato, C.J.; Dadze, A.; Rudnik-Schöneborn, S.; Simard, L.R.; Zerres, K.; Burghes, A.H. Allelic Association and Deletions in Autosomal Recessive Proximal Spinal Muscular Atrophy: Association of Marker Genotype with Disease Severity and Candidate CDNAs. Hum. Mol. Genet. 1995, 4, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Burlet, P.; Bürglen, L.; Clermont, O.; Lefebvre, S.; Viollet, L.; Munnich, A.; Melki, J. Large Scale Deletions of the 5q13 Region Are Specific to Werdnig-Hoffmann Disease. J. Med. Genet. 1996, 33, 281–283. [Google Scholar] [CrossRef]
- Velasco, E.; Valero, C.; Valero, A.; Moreno, F.; Hernández-Chico, C. Molecular Analysis of the SMN and NAIP Genes in Spanish Spinal Muscular Atrophy (SMA) Families and Correlation between Number of Copies of CBCD541 and SMA Phenotype. Hum. Mol. Genet. 1996, 5, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.R.; Owen, N.; Talbot, K.; Patel, S.; Muntoni, F.; Ignatius, J.; Dubowitz, V.; Davies, K.E. Gene Deletions in Spinal Muscular Atrophy. J. Med. Genet. 1996, 33, 93–96. [Google Scholar] [CrossRef]
- Bürglen, L.; Seroz, T.; Miniou, P.; Lefebvre, S.; Burlet, P.; Munnich, A.; Pequignot, E.V.; Egly, J.M.; Melki, J. The Gene Encoding P44, a Subunit of the Transcription Factor TFIIH, Is Involved in Large-Scale Deletions Associated with Werdnig-Hoffmann Disease. Am. J. Hum. Genet. 1997, 60, 72–79. [Google Scholar]
- Carter, T.A.; Bönnemann, C.G.; Wang, C.H.; Obici, S.; Parano, E.; De Fatima Bonaldo, M.; Ross, B.M.; Penchaszadeh, G.K.; Mackenzie, A.; Soares, M.B.; et al. A Multicopy Transcription-Repair Gene, BTF2p44, Maps to the SMA Region and Demonstrates SMA Associated Deletions. Hum. Mol. Genet. 1997, 6, 229–236. [Google Scholar] [CrossRef][Green Version]
- Ahn, E.J.; Yum, M.S.; Kim, E.H.; Yoo, H.W.; Lee, B.H.; Kim, G.H.; Ko, T.S. Genotype-Phenotype Correlation of SMN1 and NAIP Deletions in Korean Patients with Spinal Muscular Atrophy. J. Clin. Neurol. 2017, 13, 27–31. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy: A Timely Review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef]
- Hauke, J.; Riessland, M.; Lunke, S.; Eyüpoglu, I.Y.; Blümcke, I.; El-Osta, A.; Wirth, B.; Hahnen, E. Survival Motor Neuron Gene 2 Silencing by DNA Methylation Correlates with Spinal Muscular Atrophy Disease Severity and Can Be Bypassed by Histone Deacetylase Inhibition. Hum. Mol. Genet. 2009, 18, 304–317. [Google Scholar] [CrossRef]
- Kolb, S.J.; Battle, D.J.; Dreyfuss, G. Molecular Functions of the SMN Complex. J. Child Neurol. 2007, 22, 990–994. [Google Scholar] [CrossRef]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal Muscular Atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef]
- Bowerman, M.; Becker, C.G.; Yáñez-Muñoz, R.J.; Ning, K.; Wood, M.J.A.; Gillingwater, T.H.; Talbot, K. Therapeutic Strategies for Spinal Muscular Atrophy: SMN and Beyond. Dis. Model. Mech. 2017, 10, 943–954. [Google Scholar] [CrossRef]
- Stabley, D.L.; Harris, A.W.; Holbrook, J.; Chubbs, N.J.; Lozo, K.W.; Crawford, T.O.; Swoboda, K.J.; Funanage, V.L.; Wang, W.; Mackenzie, W.; et al. SMN1 and SMN2 Copy Numbers in Cell Lines Derived from Patients with Spinal Muscular Atrophy as Measured by Array Digital PCR. Mol. Genet. Genom. Med. 2015, 3, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E.R. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Seo, J.; Singh, N.N.; Singh, R.N. A Multilayered Control of the Human Survival Motor Neuron Gene Expression by Alu Elements. Front. Microbiol. 2017, 8, 2252. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.E.; Cunningham, D.; Chandler, D.S. SMN Regulation in SMA and in Response to Stress: New Paradigms and Therapeutic Possibilities. Hum. Genet. 2017, 136, 1173–1191. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Perenthaler, E.; Courtney, N.L.; Jordan, C.Y.; Shorrock, H.K.; van der Hoorn, D.; Huang, Y.-T.; Murray, L.M.; Viero, G.; Gillingwater, T.H. Temporal and Tissue-Specific Variability of SMN Protein Levels in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2018, 27, 2851–2862. [Google Scholar] [CrossRef]
- Singh, R.N.; Seo, J.; Singh, N.N. RNA in Spinal Muscular Atrophy: Therapeutic Implications of Targeting. Expert Opin. Ther. Targets 2020, 24, 731–743. [Google Scholar] [CrossRef]
- Cho, S.; Dreyfuss, G. A Degron Created by SMN2 Exon 7 Skipping Is a Principal Contributor to Spinal Muscular Atrophy Severity. Genes Dev. 2010, 24, 438–442. [Google Scholar] [CrossRef]
- Berciano, M.T.; Puente-Bedia, A.; Medina-Samamé, A.; Rodríguez-Rey, J.C.; Calderó, J.; Lafarga, M.; Tapia, O. Nusinersen Ameliorates Motor Function and Prevents Motoneuron Cajal Body Disassembly and Abnormal Poly(A) RNA Distribution in a SMA Mouse Model. Sci. Rep. 2020, 10, 10738. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, Y.; Lorson, C.L.; Stamm, S.; Androphy, E.J.; Wirth, B. Htra2-Beta 1 Stimulates an Exonic Splicing Enhancer and Can Restore Full-Length SMN Expression to Survival Motor Neuron 2 (SMN2). Proc. Natl. Acad. Sci. USA 2000, 97, 9618–9623. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.D.; Havens, M.A.; Jodelka, F.M.; Hastings, M.L. Targeting SR Proteins Improves SMN Expression in Spinal Muscular Atrophy Cells. PLoS ONE 2014, 9, e115205. [Google Scholar] [CrossRef] [PubMed]
- Pedrotti, S.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Stamm, S.; Manley, J.L.; Sette, C. The Splicing Regulator Sam68 Binds to a Novel Exonic Splicing Silencer and Functions in SMN2 Alternative Splicing in Spinal Muscular Atrophy. EMBO J. 2010, 29, 1235–1247. [Google Scholar] [CrossRef]
- Singh, N.N.; Howell, M.D.; Androphy, E.J.; Singh, R.N. How the Discovery of ISS-N1 Led to the First Medical Therapy for Spinal Muscular Atrophy. Gene Ther. 2017, 24, 520–526. [Google Scholar] [CrossRef]
- Simard, M.J.; Chabot, B. SRp30c Is a Repressor of 3’ Splice Site Utilization. Mol. Cell. Biol. 2002, 22, 4001–4010. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA Targets and Position-Dependent Splicing Regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Garzia, A.; Mazzola, M.; Gerstberger, S.; Molina, H.; Tuschl, T. The TIA1 RNA-Binding Protein Family Regulates EIF2AK2-Mediated Stress Response and Cell Cycle Progression. Mol. Cell 2018, 69, 622–635.e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-H.; Chang, J.-G.; Lu, R.-M.; Peng, T.-Y.; Tarn, W.-Y. The RNA Binding Protein HnRNP Q Modulates the Utilization of Exon 7 in the Survival Motor Neuron 2 (SMN2) Gene. Mol. Cell. Biol. 2008, 28, 6929–6938. [Google Scholar] [CrossRef] [PubMed]
- Moursy, A.; Allain, F.H.-T.; Cléry, A. Characterization of the RNA Recognition Mode of HnRNP G Extends Its Role in SMN2 Splicing Regulation. Nucleic Acids Res. 2014, 42, 6659–6672. [Google Scholar] [CrossRef]
- Tang, Z.; Zhao, J.; Pearson, Z.J.; Boskovic, Z.V.; Wang, J. RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays. Molecules 2021, 26, 2263. [Google Scholar] [CrossRef]
- Singh, N.N.; Singh, R.N.; Androphy, E.J. Modulating Role of RNA Structure in Alternative Splicing of a Critical Exon in the Spinal Muscular Atrophy Genes. Nucleic Acids Res. 2007, 35, 371–389. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lopez, A.; Tessaro, F.; Jonker, H.R.A.; Wacker, A.; Richter, C.; Comte, A.; Berntenis, N.; Schmucki, R.; Hatje, K.; Petermann, O.; et al. Targeting RNA Structure in SMN2 Reverses Spinal Muscular Atrophy Molecular Phenotypes. Nat. Commun. 2018, 9, 2032. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a Critical Exon of Human Survival Motor Neuron Is Regulated by a Unique Silencer Element Located in the Last Intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef]
- Singh, R.N.; Singh, N.N. Mechanism of Splicing Regulation of Spinal Muscular Atrophy Genes. Adv. Neurobiol. 2018, 20, 31–61. [Google Scholar] [CrossRef]
- Frederiksen, S.B.; Holm, L.L.; Larsen, M.R.; Doktor, T.K.; Andersen, H.S.; Hastings, M.L.; Hua, Y.; Krainer, A.R.; Andresen, B.S. Identification of SRSF10 as a Regulator of SMN2 ISS-N1. Hum. Mutat. 2021, 42, 246–260. [Google Scholar] [CrossRef]
- Singh, N.N.; Shishimorova, M.; Cao, L.C.; Gangwani, L.; Singh, R.N. A Short Antisense Oligonucleotide Masking a Unique Intronic Motif Prevents Skipping of a Critical Exon in Spinal Muscular Atrophy. RNA Biol. 2009, 6, 341–350. [Google Scholar] [CrossRef]
- Keil, J.M.; Seo, J.; Howell, M.D.; Hsu, W.H.; Singh, R.N.; DiDonato, C.J. A Short Antisense Oligonucleotide Ameliorates Symptoms of Severe Mouse Models of Spinal Muscular Atrophy. Mol. Ther. Nucleic Acids 2014, 3, e174. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Singh, R.N. How RNA Structure Dictates the Usage of a Critical Exon of Spinal Muscular Atrophy Gene. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194403. [Google Scholar] [CrossRef]
- Singh, N.N.; Hollinger, K.; Bhattacharya, D.; Singh, R.N. An Antisense Microwalk Reveals Critical Role of an Intronic Position Linked to a Unique Long-Distance Interaction in Pre-MRNA Splicing. RNA 2010, 16, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Lawler, M.N.; Ottesen, E.W.; Upreti, D.; Kaczynski, J.R.; Singh, R.N. An Intronic Structure Enabled by a Long-Distance Interaction Serves as a Novel Target for Splicing Correction in Spinal Muscular Atrophy. Nucleic Acids Res. 2013, 41, 8144–8165. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Miyaso, H.; Okumura, M.; Kurisu, J.; Imaizumi, K. Identification of a Cis-Acting Element for the Regulation of SMN Exon 7 Splicing. J. Biol. Chem. 2002, 277, 23271–23277. [Google Scholar] [CrossRef] [PubMed]
- Osman, E.Y.; Washington, C.W.; Kaifer, K.A.; Mazzasette, C.; Patitucci, T.N.; Florea, K.M.; Simon, M.E.; Ko, C.-P.; Ebert, A.D.; Lorson, C.L. Optimization of Morpholino Antisense Oligonucleotides Targeting the Intronic Repressor Element1 in Spinal Muscular Atrophy. Mol. Ther. 2016, 24, 1592–1601. [Google Scholar] [CrossRef]
- Godfrey, C.; Desviat, L.R.; Smedsrød, B.; Piétri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery Is Key: Lessons Learnt from Developing Splice-Switching Antisense Therapies. EMBO Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense Oligonucleotides. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-Switching Antisense Oligonucleotides as Therapeutic Drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, A.; Śliwa, A.; Żarowski, M.; Jankowska, A. Molecular Basis and Therapy of Spinal Muscular Atrophy. Child Neurol. 2019, 27, 39–46. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Warych, A.; Szoszkiewicz, M. Spinal Muscular Atrophy—Onasemnogene Abeparvovec and Other Therapeutic Options. Farm. Pol. 2020, 76, 10–17. [Google Scholar] [CrossRef]
- Jedrzejowska, M.; Milewski, M.; Zimowski, J.; Borkowska, J.; Kostera-Pruszczyk, A.; Sielska, D.; Jurek, M.; Hausmanowa-Petrusewicz, I. Phenotype Modifiers of Spinal Muscular Atrophy: The Number of SMN2 Gene Copies, Deletion in the NAIP Gene and Probably Gender Influence the Course of the Disease. Acta Biochim. Pol. 2009, 56, 103–108. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense Correction of SMN2 Splicing in the CNS Rescues Necrosis in a Type III SMA Mouse Model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Gidaro, T.; Servais, L. Nusinersen Treatment of Spinal Muscular Atrophy: Current Knowledge and Existing Gaps. Dev. Med. Child Neurol. 2019, 61, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Li, Q. Nusinersen as a Therapeutic Agent for Spinal Muscular Atrophy. Yonsei Med. J. 2020, 61, 273–283. [Google Scholar] [CrossRef]
- Dabbous, O.; Maru, B.; Jansen, J.P.; Lorenzi, M.; Cloutier, M.; Guérin, A.; Pivneva, I.; Wu, E.Q.; Arjunji, R.; Feltner, D.; et al. Survival, Motor Function, and Motor Milestones: Comparison of AVXS-101 Relative to Nusinersen for the Treatment of Infants with Spinal Muscular Atrophy Type 1. Adv. Ther. 2019, 36, 1164–1176. [Google Scholar] [CrossRef]
- Spinraza (Nusinersen); Biogen Inc.: Cambridge, MA, USA, 2018; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf (accessed on 7 July 2021).
- Rao, V.K.; Kapp, D.; Schroth, M. Gene Therapy for Spinal Muscular Atrophy: An Emerging Treatment Option for a Devastating Disease. J. Manag. Care Spec. Pharm. 2018, 24, S3–S16. [Google Scholar] [CrossRef]
- Wood, M.J.A.; Talbot, K.; Bowerman, M. Spinal Muscular Atrophy: Antisense Oligonucleotide Therapy Opens the Door to an Integrated Therapeutic Landscape. Hum. Mol. Genet. 2017, 26, R151–R159. [Google Scholar] [CrossRef]
- Foead, A.; Yeo, W.Y.; Vishnumukkala, T.; Larvin, M. Rehabilitation in Spinal Muscular Atrophy. J. Int. Soc. Phys. Rehabil. Med. 2019, 2, 62. [Google Scholar] [CrossRef]
- De Wel, B.; Goosens, V.; Sobota, A.; Van Camp, E.; Geukens, E.; Van Kerschaver, G.; Jagut, M.; Claes, K.; Claeys, K.G. Nusinersen Treatment Significantly Improves Hand Grip Strength, Hand Motor Function and MRC Sum Scores in Adult Patients with Spinal Muscular Atrophy Types 3 and 4. J. Neurol. 2021, 268, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Lee, B.M.; Singh, R.N. Splicing Regulation in Spinal Muscular Atrophy by an RNA Structure Formed by Long-Distance Interactions. Ann. N. Y. Acad. Sci. 2015, 1341, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a Phase 1 Study of Nusinersen (ISIS-SMNRx) in Children with Spinal Muscular Atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef]
- Howell, M.D.; Ottesen, E.W.; Singh, N.N.; Anderson, R.L.; Singh, R.N. Gender-Specific Amelioration of SMA Phenotype upon Disruption of a Deep Intronic Structure by an Oligonucleotide. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1328–1341. [Google Scholar] [CrossRef]
- Touznik, A.; Maruyama, R.; Hosoki, K.; Echigoya, Y.; Yokota, T. LNA/DNA Mixmer-Based Antisense Oligonucleotides Correct Alternative Splicing of the SMN2 Gene and Restore SMN Protein Expression in Type 1 SMA Fibroblasts. Sci. Rep. 2017, 7, 3672. [Google Scholar] [CrossRef] [PubMed]
- Flynn, L.L.; Mitrpant, C.; Pitout, I.L.; Fletcher, S.; Wilton, S.D. Antisense Oligonucleotide-Mediated Terminal Intron Retention of the SMN2 Transcript. Mol. Ther. Nucleic Acids 2018, 11, 91–102. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.-L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen Initiated in Infants during the Presymptomatic Stage of Spinal Muscular Atrophy: Interim Efficacy and Safety Results from the Phase 2 NURTURE Study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.Y.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor Neuron Disease. SMN2 Splicing Modifiers Improve Motor Function and Longevity in Mice with Spinal Muscular Atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Taladriz-Sender, A.; Campbell, E.; Burley, G.A. Splice-Switching Small Molecules: A New Therapeutic Approach to Modulate Gene Expression. Methods 2019, 167, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 Pre-MRNA-Protein Complex Elicits Specificity for Small Molecule Splicing Modifiers. Nat. Commun. 2017, 8, 1476. [Google Scholar] [CrossRef]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef]
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam Distributes and Increases SMN Protein in Both the Central Nervous System and Peripheral Organs. Pharmacol. Res. Perspect. 2018, 6, e00447. [Google Scholar] [CrossRef] [PubMed]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [PubMed]
- Paluszczak, J. Therapeutic Targeting of Alternative Splicing. Farm. Pol. 2019, 75, 605–616. [Google Scholar] [CrossRef]
- Wang, J.; Schultz, P.G.; Johnson, K.A. Mechanistic Studies of a Small-Molecule Modulator of SMN2 Splicing. Proc. Natl. Acad. Sci. USA 2018, 115, E4604–E4612. [Google Scholar] [CrossRef]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef]
- Singh, R.N.; Ottesen, E.W.; Singh, N.N. The First Orally Deliverable Small Molecule for the Treatment of Spinal Muscular Atrophy. Neurosci. Insights 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.J.J.; Darras, B.T. Overturning the Paradigm of Spinal Muscular Atrophy as Just a Motor Neuron Disease. Pediatr. Neurol. 2020, 109, 12–19. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal Muscular Atrophy: Going beyond the Motor Neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Evrysdi (Risdiplam) FDA Approval History. Available online: https://www.drugs.com/history/evrysdi.html (accessed on 7 July 2021).
- Novartis Pharmaceuticals. An Open Label Multi-Part First-in-Human Study of Oral LMI070 in Infants with Type 1 Spinal Muscular Atrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT02268552 (accessed on 28 June 2021).
- Palacino, J.; Swalley, S.E.; Song, C.; Cheung, A.K.; Shu, L.; Zhang, X.; Van Hoosear, M.; Shin, Y.; Chin, D.N.; Keller, C.G.; et al. SMN2 Splice Modulators Enhance U1-Pre-MRNA Association and Rescue SMA Mice. Nat. Chem. Biol. 2015, 11, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Suzuki, S.; Okubo, S.; Ohuchi, K.; Takahashi, K.; Nakamura, S.; Shimazawa, M.; Fuji, K.; Hara, H. Discovery of a CNS Penetrant Small Molecule SMN2 Splicing Modulator with Improved Tolerability for Spinal Muscular Atrophy. Sci. Rep. 2020, 10, 17472. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Androphy, E.J.; Singh, R.N. In Vivo Selection Reveals Combinatorial Controls That Define a Critical Exon in the Spinal Muscular Atrophy Genes. RNA 2004, 10, 1291–1305. [Google Scholar] [CrossRef]
- Singh, R.N. Unfolding the Mystery of Alternative Splicing through a Unique Method of in Vivo Selection. Front. Biosci. J. Virtual Libr. 2007, 12, 3263–3272. [Google Scholar] [CrossRef]
- Woll, M.G.; Qi, H.; Turpoff, A.; Zhang, N.; Zhang, X.; Chen, G.; Li, C.; Huang, S.; Yang, T.; Moon, Y.-C.; et al. Discovery and Optimization of Small Molecule Splicing Modifiers of Survival Motor Neuron 2 as a Treatment for Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 6070–6085. [Google Scholar] [CrossRef]
- Zhao, X.; Feng, Z.; Ling, K.K.Y.; Mollin, A.; Sheedy, J.; Yeh, S.; Petruska, J.; Narasimhan, J.; Dakka, A.; Welch, E.M.; et al. Pharmacokinetics, Pharmacodynamics, and Efficacy of a Small-Molecule SMN2 Splicing Modifier in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2016, 25, 1885–1899. [Google Scholar] [CrossRef]
- Ratni, H.; Karp, G.M.; Weetall, M.; Naryshkin, N.A.; Paushkin, S.V.; Chen, K.S.; McCarthy, K.D.; Qi, H.; Turpoff, A.; Woll, M.G.; et al. Specific Correction of Alternative Survival Motor Neuron 2 Splicing by Small Molecules: Discovery of a Potential Novel Medicine To Treat Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 6086–6100. [Google Scholar] [CrossRef]
- Pinard, E.; Green, L.; Reutlinger, M.; Weetall, M.; Naryshkin, N.A.; Baird, J.; Chen, K.S.; Paushkin, S.V.; Metzger, F.; Ratni, H. Discovery of a Novel Class of Survival Motor Neuron 2 Splicing Modifiers for the Treatment of Spinal Muscular Atrophy. J. Med. Chem. 2017, 60, 4444–4457. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Hurley, B.; Kerrigan, R.; Shu, L.; Chin, D.N.; Shen, Y.; O’Brien, G.; Sung, M.J.; Hou, Y.; Axford, J.; et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 11021–11036. [Google Scholar] [CrossRef]
- Kletzl, H.; Marquet, A.; Günther, A.; Tang, W.; Heuberger, J.; Groeneveld, G.J.; Birkhoff, W.; Mercuri, E.; Lochmüller, H.; Wood, C.; et al. The Oral Splicing Modifier RG7800 Increases Full Length Survival of Motor Neuron 2 MRNA and Survival of Motor Neuron Protein: Results from Trials in Healthy Adults and Patients with Spinal Muscular Atrophy. Neuromuscul. Disord. NMD 2019, 29, 21–29. [Google Scholar] [CrossRef]
- Sturm, S.; Günther, A.; Jaber, B.; Jordan, P.; Al Kotbi, N.; Parkar, N.; Cleary, Y.; Frances, N.; Bergauer, T.; Heinig, K.; et al. A Phase 1 Healthy Male Volunteer Single Escalating Dose Study of the Pharmacokinetics and Pharmacodynamics of Risdiplam (RG7916, RO7034067), a SMN2 Splicing Modifier. Br. J. Clin. Pharmacol. 2019, 85, 181–193. [Google Scholar] [CrossRef]
- Konieczny, P.; Artero, R. Drosophila SMN2 Minigene Reporter Model Identifies Moxifloxacin as a Candidate Therapy for SMA. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 3021–3036. [Google Scholar] [CrossRef]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific Inhibition of Myostatin Activation Is Beneficial in Mouse Models of SMA Therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Meng, J.; Malerba, A.; Catapano, F.; Sintusek, P.; Jarmin, S.; Feng, L.; Lu-Nguyen, N.; Sun, L.; Mariot, V.; et al. Myostatin Inhibition in Combination with Antisense Oligonucleotide Therapy Improves Outcomes in Spinal Muscular Atrophy. J. Cachexia Sarcopenia Muscle 2020, 11, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Cytokinetics. A Phase 2, Double-Blind, Randomized, Placebo-Controlled, Multiple Dose Study of CK-2127107 in Two Ascending Dose Cohorts of Patients with Spinal Muscular Atrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT02644668 (accessed on 17 August 2021).
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. NMD 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Finkel, R.; Howell, R.R.; Klinger, K.; Kuntz, N.; et al. Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef]
- Tizzano, E.F.; Finkel, R.S. Spinal Muscular Atrophy: A Changing Phenotype beyond the Clinical Trials. Neuromuscul. Disord. NMD 2017, 27, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Oprea, G.E.; Kröber, S.; McWhorter, M.L.; Rossoll, W.; Müller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 Is a Protective Modifier of Autosomal Recessive Spinal Muscular Atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.-H.; Sun, J.; Krainer, A.R.; Hua, Y.; Prior, T.W. A-44G Transition in SMN2 Intron 6 Protects Patients with Spinal Muscular Atrophy. Hum. Mol. Genet. 2017, 26, 2768–2780. [Google Scholar] [CrossRef]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a Potential Biomarker for Spinal Muscular Atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and Management of Spinal Muscular Atrophy: Part 1: Recommendations for Diagnosis, Rehabilitation, Orthopedic and Nutritional Care. Neuromuscul. Disord. NMD 2018, 28, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and Management of Spinal Muscular Atrophy: Part 2: Pulmonary and Acute Care; Medications, Supplements and Immunizations; Other Organ Systems; and Ethics. Neuromuscul. Disord. NMD 2018, 28, 197–207. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aim of the Study | Mechanism of Action | Results | References | |
|---|---|---|---|---|
| 1 | Therapeutic effect of short ASO on two mouse models of SMA: healthy, adult Smn heterozygous mice containing human SMN2 and 5058-Hemi SMA mice | Blocking GCRS | Restoring the correct splicing of exon 7 and consequently the production of full-length SMN. Proving efficacy of short ASOs in pathology and expanding the range of ASO-based substances for use in SMA therapy | Keil et al. [64] 2014 |
| 2 | Variable mechanisms regulating splicing of exon 7 in SMA-patient-derived GM03813 cell line | Targeting ISS-N1 Targeting ISS-N2 | Increasing SMN level by stimulating exon 7 inclusion by sequestration of ISS-N1 Increasing SMN and Gemin2 levels with disruption of the 3′ strands of ISTL1 and ISTL2 caused with ISS-N2 blocking Long distance interactions between intron sequences are crucial in understanding the mechanism of disrupted SMA splicing. | Singh et al. [85] 2015 |
| 3 | Improvement of ASO targeting Element 1 in SMNΔ7 mouse model | Binding potential intronic splicing silencer—E1 in upstream of exon 7 SMN2 | SMN2 splicing modification to produce full-length SMN One of the compounds being tested, E1MOv11, has the potential to become a stand-alone ASO in the clinic, but it is critical to develop combination therapy with drugs that act on other SMA pathomechanisms. | Osman et al. [69] 2016 |
| 4 | Evaluation of the tolerability, safety, pharmacokinetics, and clinical efficacy of nusinersen in cohort of 28 children with type 2 and type 3 SMA aged 2–14 years | Targeting ISS-N1 | Initiating exon 7 inclusion resulting in full-length SMN expression No safety issues found with 9 mg nusinersen dose, supporting study of higher dose. | Chiriboga et al. [86] 2016 |
| 5 | ASO effect targeting deep intronic structures to restore full-length SMN expression in allele C (C/C) mice model | Targeting ISS-N2 | A small peripheral increase in SMN alleviates SMA symptoms in a gender-specific manner—restoration of peripheral SMN production has a significant impact on testicular function.Targeting deep intron sequences is effective and has great therapeutic potential, so there is a need for further research into this strategy. | Howell et al. [87] 2017 |
| 6 | Locked nucleic acid (LNA)-based antisense oligonucleotides (LNA/DNA mixmers) as therapeutic strategy using SMA patient fibroblasts | Targeting ISS-N1 | LNA/DNA mixmer-based antisense oligonucleotide may be a potential candidate for SMA therapy. | Touznik et al. [88] 2017 |
| 7 | Mechanisms influencing ASOs-induced intron retention. ASOs impact on transcript and protein expression in SMA patient fibroblasts | Targeting SMN2 exon 8 to slowing transcription | Induction of exon/intron 7 retention Model probably not useful for SMA patients. May prove beneficial in diseases in which protein repression is crucial for therapy, e. g., cancers | Flynn et al. [89] 2018 |
| 8 | Safety and efficacy of nusinersen administration in children with cohort of 126 children with SMA who had symptom onset after 6 months of age | Targeting ISS-N1 | Children with later-onset SMA showed a significant improvement in motor function after nusinersen administration compared to control group. | Mercuri et al. [90] 2018 |
| 9 | Safety and efficacy of nusinersen in the pre-symptomatic period or at the onset of symptoms in cohort of 25 children with genetically diagnosed SMA at a median follow-up of 2. 9 years | Targeting ISS-N1 | Early screening and implementation of nusinersen therapy in the presymptomatic period significantly increases the chances for successful therapy and further normal motor development of the child treated for SMA. | De Vivo et al. [91] 2019 |
| 10 | Effects of nusinersen on the behavior of Cajal bodies (CBs) in SMN∆7 mice | Targeting ISS-N1 | Improving motor function and preventing α-motoneuron loss Selective restoring of SMN expression in the spinal cord | Berciano et al. [47] 2020 |
| Aim of Study | Mechanism of Action | Results | References | |
|---|---|---|---|---|
| 1 | Identification and optimization of a pyridazine class of orally bioavailable, small molecules enhancing inclusion SMN exon 7 in mice. | Stabilization of U1 snRNP and SMN2 pre-mRNA complex Enhancing selectively the binding affinity of U1 snRNP to 5′ss. | Modification of splicing through small sequence-specific molecules can be used in various splicing-related diseases. | Palacino et al. [107] 2015 |
| 2 | Orally deliverable small molecules correcting alternative splicing of the SMN2 gene exon 7 in SMA Δ7 mice, SMA patient fibroblasts and rats | Enhancing of the U1−pre-mRNA interaction at the 5′ splice site of SMN2 intron 7. | Reduction of disease manifestations and a significant increase in the median survival time in models after tested molecules administration Supporting the development of an orally administered small molecule for the treatment of patients with SMA | Woll et al. [111] 2016 |
| 3 | SMN-C1 in the context of preclinical data for the clinic and further therapeutic development of this series of molecules for the treatment of SMA tested in SMN∆7 mice model. | Increasing the levels of spliceosomal and U7 snRNAs. Correcting RNA processing defects induced by SMN deficiency. | Lower dose SMN-C1 increases long-term survival of SMN∆7 mouse model with partially corrected phenotype. Higher dose of SMN-C1 results in increased body weight, longer survival, and in addition, improved SMN-dependent RNA processing, spinal cord histopathology, and neuromuscular junctions. | Zhao et al. [112] 2016 |
| 4 | Improvement of coumarin and isocoumarin series, optimization of the pyridopyrimidinone series in C/C-allele SMA mouse model, SMA patient fibroblasts, spinal motor neurons SMA type I and II, and patient-derived induced pluripotent stem cells. | Induction of alternative splicing of SMN2 to exon 7 inclusion. | Discovery of selective small molecules that modify alternative splicing. | Ratni et al. [113] 2016 |
| 5 | New advanced chemotype of a small molecule discovered with SMA Δ7 mice model. | Modification of SMN2 alternative splicing to increase SMN levels. | Discovery of the two orally administrated SMN2 splicing modifiers. | Pinard et al. [114] 2017 |
| 6 | Identification of a pyridazine SMN2 pre-mRNA splicing modulator and optimization to branaplam in SMNΔ7 mouse model and SMA patient fibroblasts. | Stabilization of the interaction between the spliceosome and SMN2 pre-mRNA. | Branaplam treatment increased full-length SMN RNA and protein levels and extended survival. | Cheung et al. [115] 2018 |
| 7 | SMN-C2 and SMN-C3 promoting binding FUBP1 and KHSRP to the SMN2 pre-mRNA complex in 293T cells. | SMN-C2—binding to the AGGAAG SMN2 pre-mRNA exon 7 SMN-C3—hypothetically targets a sequence of RNA on or close to exon 7 or a splicing regulatory protein or protein complex that is specific to exon 7. | Small molecules complementary to nucleic acids modulate pre-mRNA splicing and can have a therapeutic influence on SMA. Future studies should concern recognition sequence of FUBP1 and KHSRP and their contribution in splicing regulation. | Wang et al. [99] 2018 |
| 8 | Tolerance and safety testing of RG7800 in clinical trials in cohort of Male subjects aged 23–45 years, thirteen patients with SMA, aged 13–53 years. | Modification of splicing toward promoting full-length SMN expression and downregulating SMNΔ7. | RG7800 is safe and well tolerated, and that the level of SMN after oral administration increases by twofold over the baseline concentration which may be associated with future therapeutic benefits. | Kletzl et al. [116] 2019 |
| 9 | Safety, tolerability, pharmacokinetics, and pharmacodynamics of risdiplam in cohort of 25 adult males, aged 18–45 years. Itraconazole effect on the pharmacokinetics of risdiplam. | Highly specific for pre-mRNA SMN2 splicing modifier | The tested doses of risdiplam were well tolerated and safe, and produced the desired effect of increasing full-length SMN2 pre-mRNA levels CYP3A inhibitors in the form of itraconazole have little effect on the pharmacokinetics of risdiplam. | Sturm et al. [117] 2019 |
| 10 | Preclinical characterization and prospects of TEC-1 using SMAΔ7 mice and SMA patient fibroblasts. | Binding to purine-rich regions within exon 7 Interaction with the major groove of the RNA duplex generated by the 5ʹ splicing site of exon 7 and U1 snRNA17 | Low risk of acute or chronic side effects Promising for the long-term treatment of patients with SMA Potentially higher therapeutic window compared to the SMN-C series. | Ando et al. [2] 2020 |
| 11 | Drugs that boost the minigene reporter signal within the context of Drosophila motor neurons | Promoting the inclusion of SMN2 exon 7 in a dose-dependent manner | Increasing SMN and SRSF1 levels and decreasing level of hnRNP1 with moxifloxacin The effects of moxifloxacin need to be tested in murine models as a potential SMA therapy or scaffold for other variant molecules. | Konieczny and Artero [118] 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lejman, J.; Zieliński, G.; Gawda, P.; Lejman, M. Alternative Splicing Role in New Therapies of Spinal Muscular Atrophy. Genes 2021, 12, 1346. https://doi.org/10.3390/genes12091346
Lejman J, Zieliński G, Gawda P, Lejman M. Alternative Splicing Role in New Therapies of Spinal Muscular Atrophy. Genes. 2021; 12(9):1346. https://doi.org/10.3390/genes12091346
Chicago/Turabian StyleLejman, Jan, Grzegorz Zieliński, Piotr Gawda, and Monika Lejman. 2021. "Alternative Splicing Role in New Therapies of Spinal Muscular Atrophy" Genes 12, no. 9: 1346. https://doi.org/10.3390/genes12091346
APA StyleLejman, J., Zieliński, G., Gawda, P., & Lejman, M. (2021). Alternative Splicing Role in New Therapies of Spinal Muscular Atrophy. Genes, 12(9), 1346. https://doi.org/10.3390/genes12091346