
A Draft Genome of the Ginger Species Alpinia nigra and New Insights into the Genetic Basis of Flexistyly


Abstract
1. Introduction
2. Materials and Methods
2.1. Study System
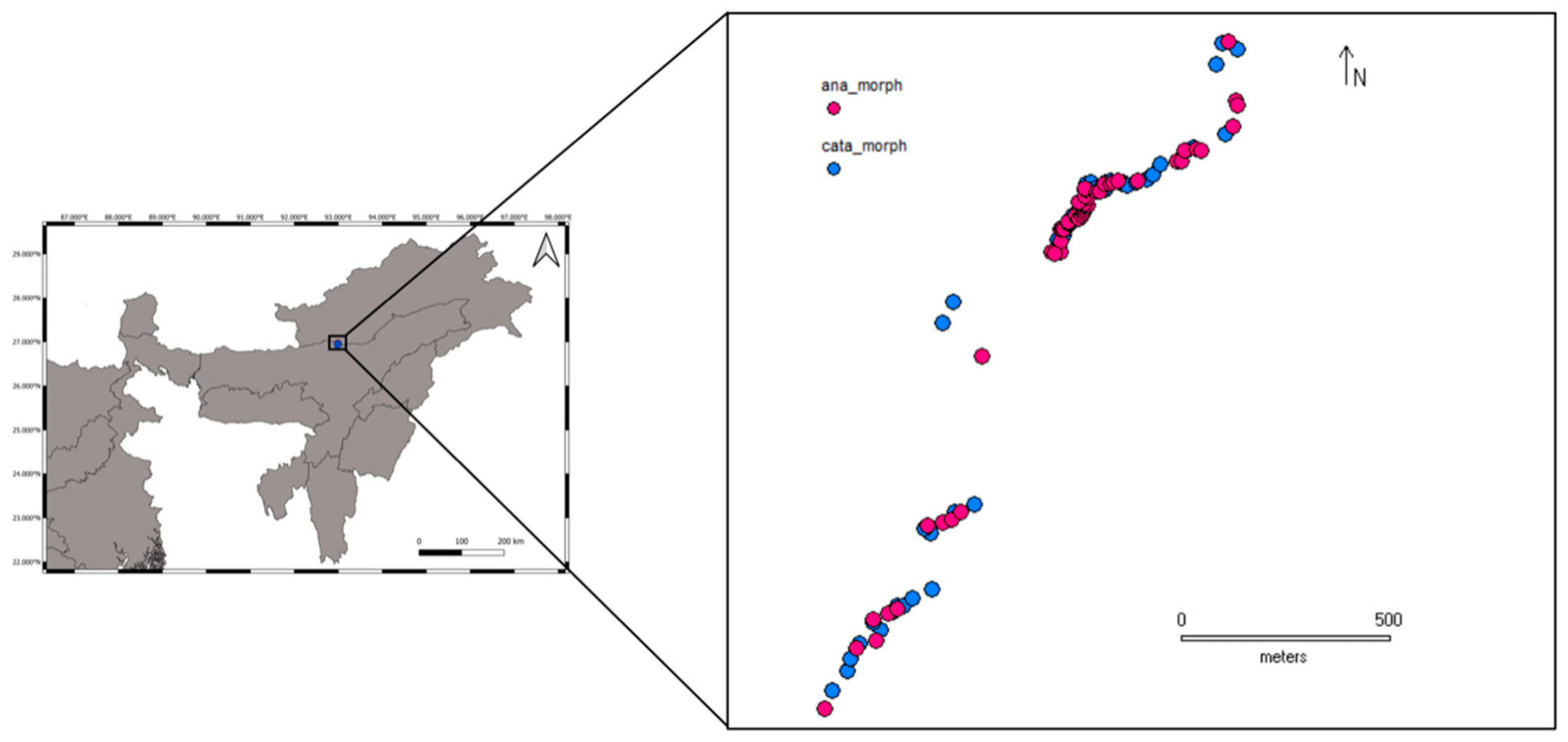
2.2. Sample Collection
2.3. Reference Genome Assembly
2.3.1. DNA Extraction (Genome)
2.3.2. Genome Assembly
2.4. Pool-Seq
2.4.1. DNA Extraction
2.4.2. Genome Mapping and Analysis
3. Results
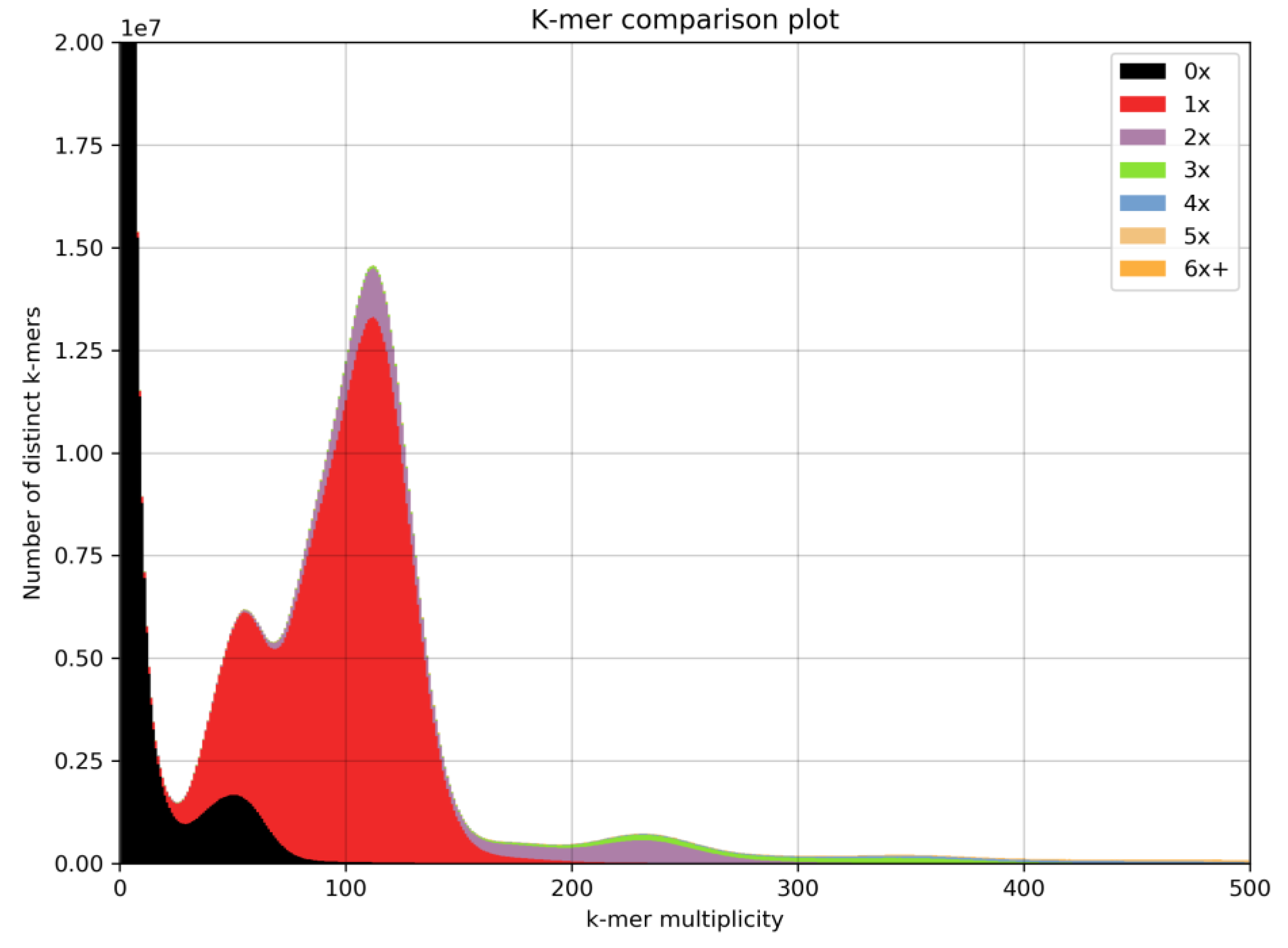
3.1. Alpinia Nigra Genome Assembly
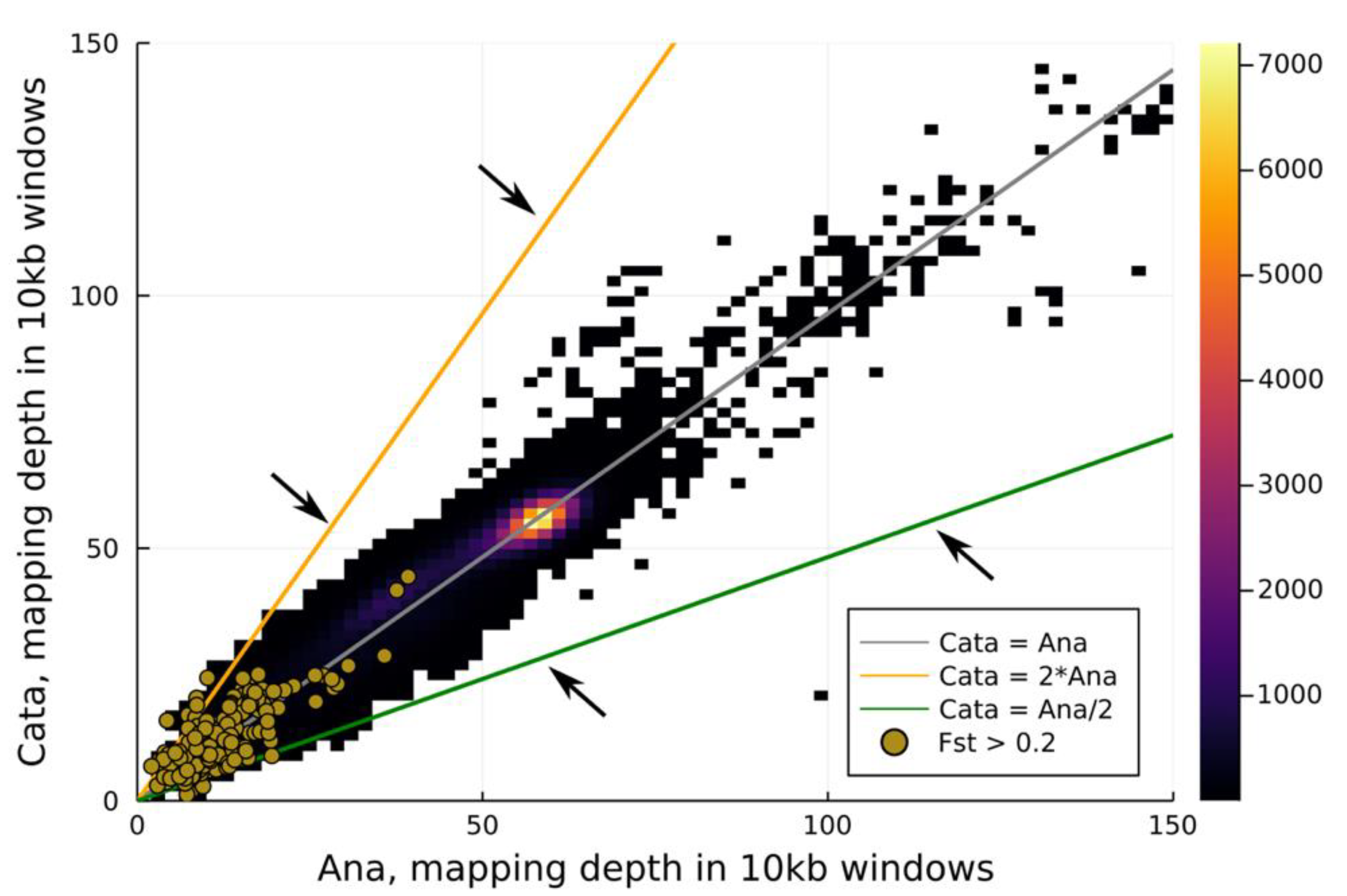
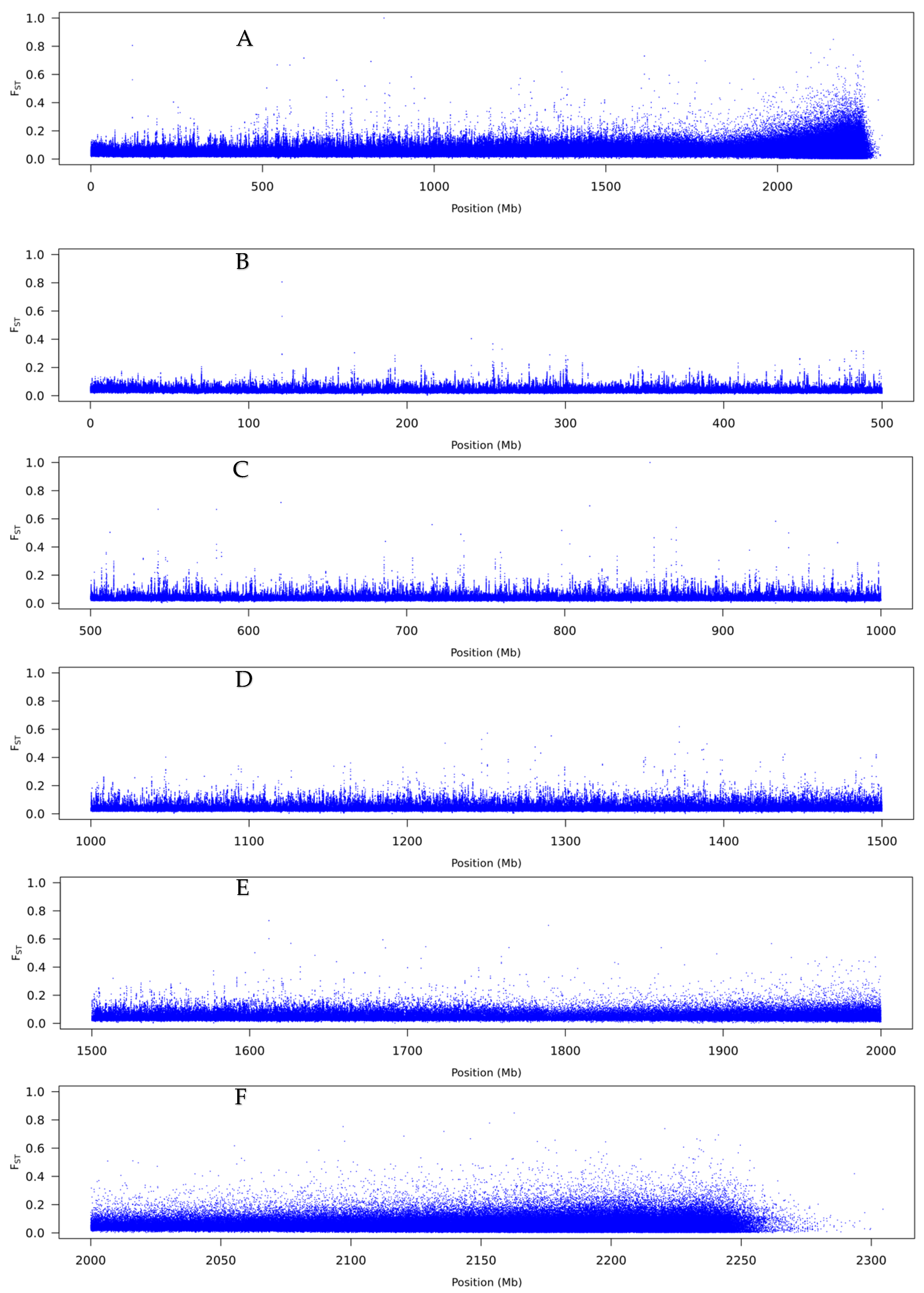
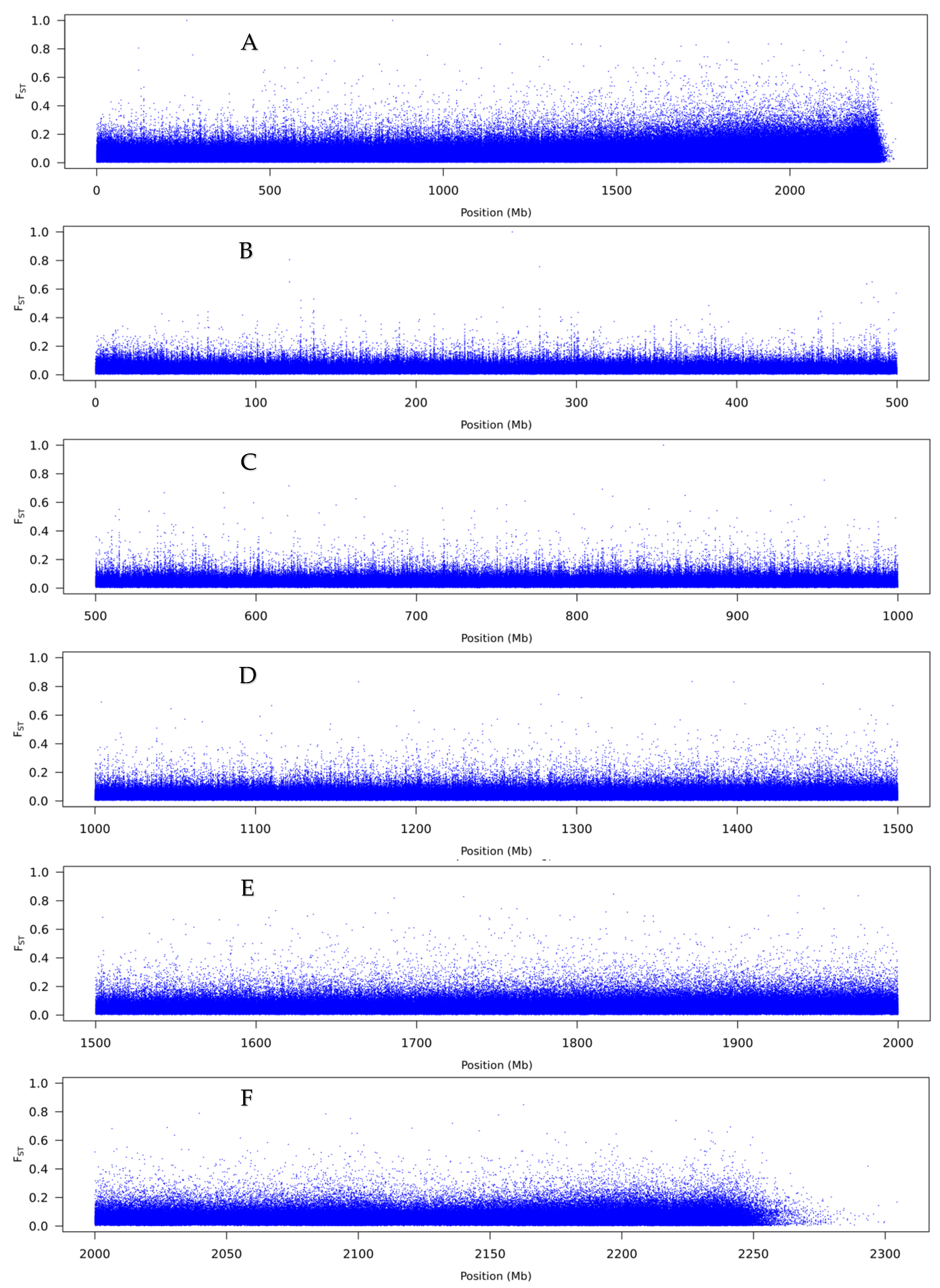
3.2. Pool-Seq
4. Discussion
4.1. Draft Genome Assembly of Alpinia nigra
4.2. The Genetic Basis of Flexistyly
4.3. The Evolution of Flexistyly
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrett, S.C.H. Understanding plant reproductive diversity. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Waser, N.M.; Ollerton, J. Geographical variation in diversity and specificity of pollination systems. In Plant-Pollinator Interactions: From Specialization to Generalization; Ollerton, J., Johnson, S.D., Hingston, A.B., Eds.; University of Chicago Press: Chicago, IL, USA, 2006; pp. 283–308. [Google Scholar]
- Barrett, S.C.H. Darwin’s legacy: The forms, function and sexual diversity of flowers. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 351–368. [Google Scholar] [CrossRef]
- Barrett, S.C.H. Sexual interference of the floral kind. Heredity 2002, 88, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D.; Charlesworth, B. Inbreeding depression and its evolutionary consequences. Annu. Rev. Ecol. Syst. 1987, 18, 237–268. [Google Scholar] [CrossRef]
- Ornduff, R. Historical Perspectives on Heterostyly. In Evolution and Function of Heterostyly. Monographs on Theoretical and Applied Genetics; Barrett, S.C.H., Ed.; Springer: Berlin/Heidelberg, Germany, 1992; Volume 15, pp. 31–39. [Google Scholar]
- Darwin, C. The Different Forms of Flowers on Plants of the Same Species; John Murray: London, UK, 1877. [Google Scholar]
- Lewis, D.; Jones, D.A. The genetics of heterostyly. In Evolution and Function of Heterostyly; Barrett, S.C.H., Ed.; Springer: New York, NY, USA, 1992; pp. 129–150. [Google Scholar]
- Charlesworth, D. The status of supergenes in the 21st century: Recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evol. Appl. 2016, 9, 74–90. [Google Scholar] [CrossRef]
- Huu, C.N.; Kappel, C.; Keller, B.; Sicard, A.; Takebayashi, Y.; Breuninger, H.; Nowak, M.D.; Baeurle, I.; Himmelbach, A.; Burkart, M. Presence versus absence of CYP734A50 underlies the style-length dimorphism in primroses. E Life 2016, 5, e17956. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cocker, J.M.; Wright, J.; Webster, M.A.; McMullan, M.; Dyer, S.; Swarbreck, D.; Caccamo, M.; van Oosterhout, C.; Gilmartin, P.M. Genetic architecture and evolution of the S locus supergene in Primula vulgaris. Nat. Plants 2016, 2, 16188. [Google Scholar] [CrossRef]
- Huu, C.N.; Keller, B.; Conti, E.; Kappel, C.; Lenhard, M. Supergene evolution via stepwise duplications and neofunctionalization of a floral-organ identity gene. Proc. Natl. Acad. Sci. USA 2020, 117, 23148–23157. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Mori, M.; Aii, J.; Abe, T.; Matsumoto, D.; Sato, S.; Hayashi, Y.; Ohnishi, O.; Ota, T. S-LOCUS EARLY FLOWERING 3 is exclusively present in the genomes of short-styled buckwheat plants that exhibit heteromorphic self-incompatibility. PLoS ONE 2012, 7, e31264. [Google Scholar] [CrossRef]
- Ushijima, K.; Nakano, R.; Bando, M.; Shigezane, Y.; Ikeda, K.; Namba, Y.; Kume, S.; Kitabata, T.; Mori, H.; Kubo, Y. Isolation of the floral morph-related genes in heterostylous flax (Linum grandiflorum): The genetic polymorphism and the transcriptional and post-transcriptional regulations of the S locus. Plant J. 2012, 69, 317–331. [Google Scholar] [CrossRef]
- Shore, J.S.; Hamam, H.J.; Chafe, P.D.J.; Labonne, J.D.J.; Henning, P.M.; McCubbin, A.G. The long and short of the S-locus in Turnera (Passifloraceae). New Phytol. 2019, 224, 1316–1329. [Google Scholar] [CrossRef]
- Li, Q.J.; Xu, Z.F.; Kress, W.J.; Xia, Y.M.; Zhang, L.; Deng, X.B.; Gao, J.Y.; Bai, Z.L. Flexible style that encourages outcrossing. Nature 2001, 410, 432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Q.J.; Deng, X.B.; Ren, P.Y.; Gao, J.Y. Reproductive biology of Alpinia blepharocalyx (Zingiberaceae): Another example of flexistyly. Plant Syst. Evol. 2003, 241, 67–76. [Google Scholar] [CrossRef]
- Li, Q.J.; Kress, W.J.; Xu, Z.-F.; Xia, Y.-M.; Zhang, L.; Deng, X.-B.; Gao, J.Y. Mating system and stigmatic behaviour during flowering of Alpinia kwangsiensis (Zingiberaceae). Plant Syst. Evol. 2002, 232, 123–132. [Google Scholar] [CrossRef]
- Renner, S.S. How common is heterodichogamy? Trends Ecol. Evol. 2001, 16, 595–597. [Google Scholar] [CrossRef]
- Sun, S.; Gao, J.Y.; Liao, W.J.; Li, Q.J.; Zhang, D.Y. Adaptive significance of flexistyly in Alpinia blepharocalyx (Zingiberaceae): A hand-pollination experiment. Ann. Bot. 2007, 99, 661–666. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cui, X.; Wei, R.; Huang, R. A study on the breeding system of Amomum tsao-ko. In Proceedings of the Second Symposium on the Family Zingiberaceae; Wu, T.L., Wu, Q.G., Chen, Z.Y., Eds.; South China Institute of Botany: Guangzhou, China, 1996; pp. 288–296. [Google Scholar]
- Kress, W.J.; Liu, A.-Z.; Newman, M.; Li, Q.J. The molecular phylogeny of Alpinia (Zingiberaceae): A complex and polyphyletic genus of gingers. Am. J. Bot. 2005, 92, 167–178. [Google Scholar] [CrossRef]
- Jia, X.-C.; Li, J.; Lu, G.-H.; Wang, Y.-Q. The concrete evidence of flexistyly in Plagiostachys: Pollination biology of a wild ginger on Hainan Island, China. Ecol. Evol. 2015, 5, 5364–5371. [Google Scholar] [CrossRef]
- Leong-Škorničková, J.; Tran, H.Ð.; Nguyen, Q.B.; Šída, O. Siliquamomum alcicorne (Zingiberaceae: Alpinioideae), a new species from central Vietnam. Gard. Bull. 2014, 66, 39–46. [Google Scholar]
- Poulsen, A.D.; Phonsena, P. Morphological variation and distribution of the useful ginger Etlingera pavieana (Zingiberaceae). Nord. J. Bot. 2017, 35, 467–475. [Google Scholar] [CrossRef]
- Gleeson, S.K. Heterodichogamy in walnuts: Inheritance and stable ratios. Evolution 1982, 36, 892. [Google Scholar] [CrossRef]
- Thompson, T.E.; Romberg, L.D. Inheritance of heterodichogamy in pecan. J. Hered. 1985, 76, 456–458. [Google Scholar] [CrossRef]
- Futschik, A.; Schlötterer, C. The next generation of molecular markers from massively parallel sequencing of pooled DNA samples. Genetics 2010, 186, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Schlötterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals—Mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Mahajan, S.; Jaiswal, S.K.; Sharma, V.K. Genome sequencing of turmeric provides evolutionary insights into its medicinal properties. bioRxiv 2020. [Google Scholar] [CrossRef]
- D’Hont, A.; Denoeud, F.; Aury, J.-M.; Baurens, F.-C.; Carreel, F.; Garsmeur, O.; Noel, B.; Bocs, S.; Droc, G.; Rouard, M.; et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012, 488, 213–217. [Google Scholar] [CrossRef]
- Davey, M.W.; Gudimella, R.; Harikrishna, J.A.; Sin, L.W.; Khalid, N.; Keulemans, J. A draft Musa balbisiana genome sequence for molecular genetics in polyploid, inter- and intra-specific Musa hybrids. BMC Genom. 2013, 14, 683. [Google Scholar] [CrossRef]
- Wu, W.; Yang, Y.-L.; He, W.-M.; Rouard, M.; Li, W.-M.; Xu, M.; Roux, N.; Ge, X.-J. Whole genome sequencing of a banana wild relative Musa itinerans provides insights into lineage-specific diversification of the Musa genus. Sci. Rep. 2016, 6, 31586. [Google Scholar] [CrossRef]
- Belser, C.; Istace, B.; Denis, E.; Dubarry, M.; Baurens, F.-C.; Falentin, C.; Genete, M.; Berrabah, W.; Chèvre, A.-M.; Delourme, R.; et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 2018, 4, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Lowry, D.B.; Willis, J.H. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol. 2010, 8, e1000500. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Pyhäjärvi, T.; Weber, A.L.; Dawe, R.K.; Glaubitz, J.C.; González, J.d.J.S.; Ross-Ibarra, C.; Doebley, J.; Morrell, P.L.; Ross-Ibarra, J. Megabase-scale inversion polymorphism in the wild ancestor of maize. Genetics 2012, 191, 883–894. [Google Scholar] [CrossRef]
- Hooper, D.M.; Price, T.D. Chromosomal inversion differences correlate with range overlap in passerine birds. Nat. Ecol. Evol. 2017, 1, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Joron, M.; Frezal, L.; Jones, R.T.; Chamberlain, N.L.; Lee, S.F.; Haag, C.R.; Whibley, A.; Becuwe, M.; Baxter, S.W.; Ferguson, L.; et al. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 2011, 477, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.M. Alpinia (Zingiberaceae): A proposed new infrageneric classification. Edinb. J. Bot. 1990, 47, 1–75. [Google Scholar] [CrossRef]
- Ma, X.-N.; Xie, C.-L.; Miao, Z.; Yang, Q.; Yang, X.-W. An overview of chemical constituents from Alpinia species in the last six decades. RSC Adv. 2017, 7, 14114. [Google Scholar] [CrossRef]
- Raghavan, T.S.; Venkatasubban, K.R. Cytological studies in the family Zingiberaceae with special reference to chromosome number and Cyto-Taxonomy. Proc. Indian Acad. Sci.-Sect. B 1943, 17, 118–132. [Google Scholar] [CrossRef]
- Chen, Z. Cytotaxonomy of the tribe Alpineae. In Proceedings of the Second Symposium on the Family Zingiberaceae; Zhongshan University Press: Guangzhou, China, 1996; pp. 112–121. [Google Scholar]
- Basak, S.; Rangan, L. New record of nuclear DNA amounts of some Zingiberaceae species from North east India. Data Br. 2018, 17, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, P.; Poulsen, A.D.; Harris, D.; Forrest, L.L. The collection and storage of plant material for DNA extraction: The Teabag Method. Gard. Bull. Singap. 2013, 65, 231–234. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 2 June 2021).
- Mapleson, D.; Garcia Accinelli, G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 2016, 33, 574–576. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Yin, S.; Sharpe, T.; Lau, B.; Hegarty, R.; Holmes, L.; Sogoloff, B.; Tabbaa, D.; Williams, L.; Russ, C.; et al. Comprehensive variation discovery in single human genomes. Nat. Genet. 2014, 46, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of genome assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Kofler, R.; Pandey, R.V.; Schlötterer, C. PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 2011, 27, 3435–3436. [Google Scholar] [CrossRef]
- Doležel, J.; Bartoš, J.; Voglmayr, H.; Greilhuber, J. Letter to the editor. Cytom. Part A 2003, 51A, 127–128. [Google Scholar] [CrossRef]
- Love, R.R.; Weisenfeld, N.I.; Jaffe, D.B.; Besansky, N.J.; Neafsey, D.E. Evaluation of DISCOVAR de novo using a mosquito sample for cost-effective short-read genome assembly. BMC Genom. 2016, 17, 187. [Google Scholar] [CrossRef]
- Tamiru, M.; Natsume, S.; Takagi, H.; White, B.; Yaegashi, H.; Shimizu, M.; Yoshida, K.; Uemura, A.; Oikawa, K.; Abe, A.; et al. Genome sequencing of the staple food crop white Guinea yam enables the development of a molecular marker for sex determination. BMC Biol. 2017, 15, 86. [Google Scholar] [CrossRef]
- Emmrich, P.M.F.; Sarkar, A.; Njaci, I.; Kaithakottil, G.G.; Ellis, N.; Moore, C.; Edwards, A.; Heavens, D.; Waite, D.; Cheema, J.; et al. A draft genome of grass pea (Lathyrus sativus), a resilient diploid legume. bioRxiv 2020. [Google Scholar] [CrossRef]
- Clavijo, B.J.; Venturini, L.; Schudoma, C.; Accinelli, G.G.; Kaithakottil, G.; Wright, J.; Borrill, P.; Kettleborough, G.; Heavens, D.; Chapman, H.; et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017, 27, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.; Batley, J. Plant genome sequencing: Applications for crop improvement. Plant Biotechnol. J. 2010, 8, 2–9. [Google Scholar] [CrossRef]
- Lichman, B.R.; Godden, G.T.; Hamilton, J.P.; Palmer, L.; Kamileen, M.O.; Zhao, D.; Vaillancourt, B.; Wood, J.C.; Sun, M.; Kinser, T.J.; et al. The evolutionary origins of the cat attractant nepetalactone in catnip. Sci. Adv. 2020, 6, eaba0721. [Google Scholar] [CrossRef] [PubMed]
- Thomma, B.P.H.J.; Seidl, M.F.; Shi-Kunne, X.; Cook, D.E.; Bolton, M.D.; van Kan, J.A.L.; Faino, L. Mind the gap; seven reasons to close fragmented genome assemblies. Fungal Genet. Biol. 2016, 90, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Wu, D.; Li, G.; Wu, D.; Wang, Z. Next-generation sequencing from bulked segregant analysis identifies a dwarfism gene in watermelon. Sci. Rep. 2018, 8, 2908. [Google Scholar] [CrossRef] [PubMed]
- Gould, B.A.; Chen, Y.; Lowry, D.B. Pooled ecotype sequencing reveals candidate genetic mechanisms for adaptive differentiation and reproductive isolation. Mol. Ecol. 2017, 26, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Kurland, S.; Wheat, C.W.; Paz Celorio Mancera, M.; Kutschera, V.E.; Hill, J.; Andersson, A.; Rubin, C.; Andersson, L.; Ryman, N.; Laikre, L. Exploring a Pool-seq-only approach for gaining population genomic insights in nonmodel species. Ecol. Evol. 2019, 9, 11448–11463. [Google Scholar] [CrossRef]
- Ren, P.Y.; Liu, M.; Li, Q.J. An example of flexistyly in a wild cardamom species (Amomum maximum, (Zingiberaceae)). Plant Syst. Evol. 2007, 267, 147–154. [Google Scholar] [CrossRef]
- Jay, P.; Whibley, A.; Frézal, L.; Rodríguez de Cara, M.Á.; Nowell, R.W.; Mallet, J.; Dasmahapatra, K.K.; Joron, M. Supergene evolution triggered by the introgression of a chromosomal inversion. Curr. Biol. 2018, 28, 1839–1845.e3. [Google Scholar] [CrossRef]
- Twyford, A.D.; Friedman, J. Adaptive divergence in the monkey flower Mimulus guttatus is maintained by a chromosomal inversion. Evolution 2015, 69, 1476–1486. [Google Scholar] [CrossRef]
- Delprat, A.; Negre, B.; Puig, M.; Ruiz, A. The transposon Galileo generates natural chromosomal inversions in Drosophila by ectopic recombination. PLoS ONE 2009, 4, e7883. [Google Scholar] [CrossRef]
- Cocker, J.M.; Wright, J.; Li, J.; Swarbreck, D.; Dyer, S.; Caccamo, M.; Gilmartin, P.M. Primula vulgaris (primrose) genome assembly, annotation and gene expression, with comparative genomics on the heterostyly supergene. Sci. Rep. 2018, 8, 17942. [Google Scholar] [CrossRef]
- Wellenreuther, M.; Bernatchez, L. Eco-evolutionary genomics of chromosomal inversions. Trends Ecol. Evol. 2018, 33, 427–440. [Google Scholar] [CrossRef]
- Thompson, N.F.; Anderson, E.C.; Clemento, A.J.; Campbell, M.A.; Pearse, D.E.; Hearsey, J.W.; Kinziger, A.P.; Garza, J.C. A complex phenotype in salmon controlled by a simple change in migratory timing. Science 2020, 370, 609–613. [Google Scholar] [CrossRef]
- Wang, J.; Ding, J.; Tan, B.; Robinson, K.M.; Michelson, I.H.; Johansson, A.; Nystedt, B.; Scofield, D.G.; Nilsson, O.; Jansson, S.; et al. A major locus controls local adaptation and adaptive life history variation in a perennial plant. Genome Biol. 2018, 19, 72. [Google Scholar] [CrossRef]
- Luo, Y.-L.; Bi, T.-J.; Li, D.; Su, Z.-L.; Tao, C.; Luo, Y.-J.; Li, Q.-J. Effects of indole-3-acetic acid and auxin transport inhibitors on the style curvature of three Alpinia species (Zingiberaceae). Acta Physiol. Plant. 2012, 34, 2019–2025. [Google Scholar] [CrossRef]
- Luo, Y.-L.; Li, Q.-J. Effects of light and low temperature on the reciprocal style curvature of flexistylous Alpinia species (Zingiberaceae). Acta Physiol. Plant. 2010, 32, 1229–1234. [Google Scholar] [CrossRef]
- Su, Z.-L.; Cui, X.-L.; Li, L.; Li, Q.-J.; Luo, Y.-L. Similar behavior, different mechanisms: The research of the style bending of Alpinia species. Acta Physiol. Plant. 2017, 39, 143. [Google Scholar] [CrossRef]
- Primack, R.B. Longevity of Individual Flowers. Annu. Rev. Ecol. Syst. 1985, 16, 15–37. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, D.Y.; Ives, A.R.; Li, Q.J. Why do stigmas move in a flexistylous plant? J. Evol. Biol. 2011, 24, 497–504. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Length (≥0 bp) | 2389 Mb |
|---|---|
| Total length (≥1000 bp) | 2119 Mb |
| No. of contigs(≥0 bp) | 1,072,070 |
| No. of contigs (≥1000 bp) | 158,389 |
| N50 Length | 48.9 Kb |
| Longest contig | 591.2 Kb |
| BUSCO score 1 | C: 91.1%, (S: 84.1%, D: 7.0%), F: 4.9%, M: 4.0%, n: 1375 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranavat, S.; Becher, H.; Newman, M.F.; Gowda, V.; Twyford, A.D. A Draft Genome of the Ginger Species Alpinia nigra and New Insights into the Genetic Basis of Flexistyly. Genes 2021, 12, 1297. https://doi.org/10.3390/genes12091297
Ranavat S, Becher H, Newman MF, Gowda V, Twyford AD. A Draft Genome of the Ginger Species Alpinia nigra and New Insights into the Genetic Basis of Flexistyly. Genes. 2021; 12(9):1297. https://doi.org/10.3390/genes12091297
Chicago/Turabian StyleRanavat, Surabhi, Hannes Becher, Mark F. Newman, Vinita Gowda, and Alex D. Twyford. 2021. "A Draft Genome of the Ginger Species Alpinia nigra and New Insights into the Genetic Basis of Flexistyly" Genes 12, no. 9: 1297. https://doi.org/10.3390/genes12091297
APA StyleRanavat, S., Becher, H., Newman, M. F., Gowda, V., & Twyford, A. D. (2021). A Draft Genome of the Ginger Species Alpinia nigra and New Insights into the Genetic Basis of Flexistyly. Genes, 12(9), 1297. https://doi.org/10.3390/genes12091297