
Identification of Copy Number Variants in a Southern Chinese Cohort of Patients with Congenital Scoliosis

 , , ,
, , ,  and
and
Abstract
:1. Introduction
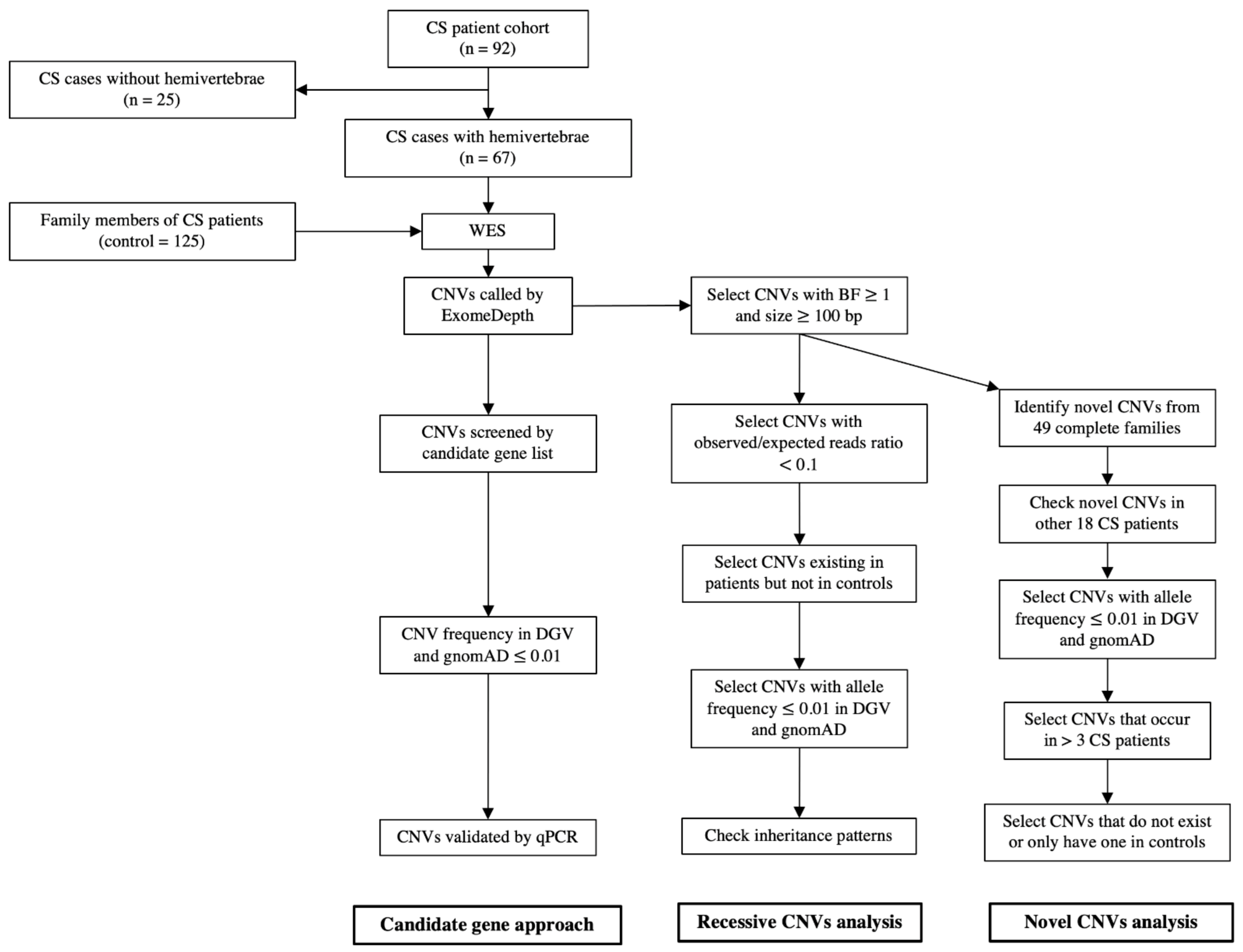
2. Materials and Methods
2.1. Patient Recruitment
2.2. Control Cohort
2.3. Genomic DNA Extraction
2.4. Whole-Exome Sequencing (WES) and Copy Number Variations (CNVs) Calling
2.5. CNVs Filtering
2.6. Real-Time Quantitative PCR (qPCR)
3. Results
3.1. CS Cohort and WES
3.2. CNV Calling
3.3. CNVs in Candidate Genes
3.4. Recessive CNVs in Patients with CS
3.5. Novel CNVs in Patients with CS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giampietro, P.F. Genetic Aspects of Congenital and Idiopathic Scoliosis. Scientifica 2012, 2012, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Shands, A.R.; Eisberg, H.B. The incidence of scoliosis in the state of Delaware; a study of 50,000 minifilms of the chest made during a survey for tuberculosis. J. Bone Jt. Surg. Am. Vol. 1955, 37, 1243–1249. [Google Scholar] [CrossRef]
- Wynne-Davies, R. Congenital vertebral anomalies: Aetiology and relationship to spina bifida cystica. J. Med. Genet. 1975, 12, 280–288. [Google Scholar] [CrossRef] [Green Version]
- McMaster, M.J.; Ohtsuka, K. The natural history of congenital scoliosis. A study of two hundred and fifty-one patients. J. Bone Jt. Surg. Am. Vol. 1982, 64, 1128–1147. [Google Scholar] [CrossRef]
- McMaster, M.J.; Singh, H. Natural History of Congenital Kyphosis and Kyphoscoliosis. A Study of One Hundred and Twelve Patients. J. Bone Jt. Surg. Am. Vol. 1999, 81, 1367–1383. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Castelein, R.M.; Chu, W.C.; Danielsson, A.J.; Dobbs, M.B.; Grivas, T.B.; Gurnett, C.A.; Luk, K.D.; Moreau, A.; Newton, P.O.; et al. Adolescent idiopathic scoliosis. Nat. Rev. Dis. Primers 2015, 1, 15030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourquié, O. Vertebrate Segmentation: From Cyclic Gene Networks to Scoliosis. Cell 2011, 145, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Maroto, M.; Bone, R.A.; Dale, J.K. Somitogenesis. Development 2012, 139, 2453–2456. [Google Scholar] [CrossRef] [Green Version]
- Giampietro, P.; Raggio, C.; Blank, R.D.; McCarty, C.; Broeckel, U.; Pickart, M. Clinical, Genetic and Environmental Factors Associated with Congenital Vertebral Malformations. Mol. Syndr. 2012, 4, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Månér, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-Scale Copy Number Polymorphism in the Human Genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.G.; Alvarado, D.M.; Haller, G.; Aferol, H.; Miller, N.H.; Dobbs, M.B.; Gurnett, C.A. Are copy number variants associated with adolescent idiopathic scoliosis? Clin. Orthop. Relat. Res. 2014, 472, 3216–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadler, B.; Haller, G.; Antunes, L.; Bledsoe, X.; Morcuende, J.; Giampietro, P.; Raggio, C.; Miller, N.; Kidane, Y.; Wise, C.A.; et al. Distal chromosome 16p11.2 duplications containing SH2B1 in patients with scoliosis. J. Med. Genet. 2019, 56, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Al-Kateb, H.; Khanna, G.; Filges, I.; Hauser, N.; Grange, D.K.; Shen, J.; Smyser, C.D.; Kulkarni, S.; Shinawi, M. Scoliosis and vertebral anomalies: Additional abnormal phenotypes associated with chromosome 16p11.2 rearrangement. Am. J. Med. Genet. Part A 2014, 164, 1118–1126. [Google Scholar] [CrossRef]
- Wu, N.; Ming, X.; Xiao, J.; Wu, Z.; Chen, X.; Shinawi, M.; Shen, Y.; Yu, G.; Liu, J.; Xie, H.; et al. TBX6 Null Variants and a Common Hypomorphic Allele in Congenital Scoliosis. N. Engl. J. Med. 2015, 372, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Kou, I.; Kawakami, N.; Iida, A.; Nakajima, M.; Ogura, Y.; Imagawa, E.; Miyake, N.; Matsumoto, N.; Yasuhiko, Y.; et al. Compound Heterozygosity for Null Mutations and a Common Hypomorphic Risk Haplotype in TBX6 Causes Congenital Scoliosis. Hum. Mutat. 2017, 38, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, M.; Duffourd, Y.; Jouan, T.; Poe, C.; Jean-Marçais, N.; Verloes, A.; St-Onge, J.; Riviere, J.-B.; Petit, F.; Pierquin, G.; et al. Autosomal recessive variations of TBX6, from congenital scoliosis to spondylocostal dysostosis. Clin. Genet. 2016, 91, 908–912. [Google Scholar] [CrossRef]
- Feng, X.; Cheung, J.P.Y.; Je, J.S.H.; Cheung, P.W.H.; Chen, S.; Yue, M.; Wang, N.; Choi, V.N.T.; Yang, X.; Song, Y.; et al. Genetic variants of TBX6 and TBXT identified in patients with congenital scoliosis in Southern China. J. Orthop. Res. 2020, 39, 971–988. [Google Scholar] [CrossRef]
- Liu, Z.L.; Wu, N.; Wu, Z.H.; Zuo, Y.Z.; Qiu, G.X. Advances in congenital vertebral malformation caused by genomic copy number variation. Zhonghua Wai Ke Za Zhi 2016, 54, 313–316. [Google Scholar]
- Gao, X.; Gotway, G.; Rathjen, K.; Johnston, C.; Sparagana, S.; Wise, C.A. Genomic Analyses of Patients with Unexplained Early-Onset Scoliosis. Spine Deform. 2014, 2, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, N.; Schaffer, A.A.; Liu, X.; Zhao, Z.; Elliott, G.; Garrett, L.; Choi, N.T.; Wang, Y.; Wang, Y.; et al. Mutations in COMP cause familial carpal tunnel syndrome. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, R.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.; Stebbings, E.; Grigoriadou, S.; Wood, N.; Hambleton, S.; Burns, S.; Thrasher, A.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Burnei, G.; Gavriliu, S.; Vlad, C.; Georgescu, I.; Ghita, R.A.; Dughilă, C.; Japie, E.M.; Onilă, A. Congenital scoliosis: An up-to-date. J. Med. Life 2015, 8, 388–397. [Google Scholar]
- Ellingford, J.M.; Campbell, C.; Barton, S.; Bhaskar, S.; Gupta, S.; Taylor, R.L.; Sergouniotis, P.I.; Horn, B.; Lamb, J.; Michaelides, M.; et al. Validation of copy number variation analysis for next-generation sequencing diagnostics. Eur. J. Hum. Genet. 2017, 25, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Gao, X.; Londono, D.; Devroy, S.E.; Mauldin, K.N.; Frankel, J.T.; Brandon, J.M.; Zhang, D.; Li, Q.-Z.; Dobbs, M.B.; et al. Genome-wide association studies of adolescent idiopathic scoliosis suggest candidate susceptibility genes. Hum. Mol. Genet. 2011, 20, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Bashiardes, S.; Veile, R.; Allen, M.; Wise, C.A.; Dobbs, M.; Morcuende, J.A.; Szappanos, L.; Herring, J.A.; Bowcock, A.M.; Lovett, M. SNTG1, the gene encoding gamma1-syntrophin: A candidate gene for idiopathic scoliosis. Hum. Genet. 2004, 115, 81–89. [Google Scholar] [CrossRef]
- Tassano, E.; Ronchetto, P.; Severino, M.; Divizia, M.; Lerone, M.; Uccella, S.; Nobili, L.; Tavella, E.; Morerio, C.; Coviello, D.; et al. Scoliosis with cognitive impairment in a girl with 8q11.21q11.23 microdeletion and SNTG1 disruption. Bone 2021, 150, 116022. [Google Scholar] [CrossRef]
- Paine, I.; Posey, J.; Grochowski, C.M.; Jhangiani, S.N.; Rosenheck, S.; Kleyner, R.; Marmorale, T.; Yoon, M.; Wang, K.; Robison, R.; et al. Paralog Studies Augment Gene Discovery: DDX and DHX Genes. Am. J. Hum. Genet. 2019, 105, 302–316. [Google Scholar] [CrossRef] [Green Version]
- Lupski, J.R. Genomic rearrangements and sporadic disease. Nat. Genet. 2007, 39, S43–S47. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, D.B.; Chapman, G.; Smith, A.J.; Mattar, M.Z.; Major, J.A.; O’Reilly, V.C.; Saga, Y.; Zackai, E.H.; Dormans, J.P.; Alman, B.A.; et al. A Mechanism for Gene-Environment Interaction in the Etiology of Congenital Scoliosis. Cell 2012, 149, 295–306. [Google Scholar] [CrossRef] [Green Version]
- McDaniell, R.; Warthen, D.M.; Sanchez-Lara, P.A.; Pai, A.; Krantz, I.D.; Piccoli, D.A.; Spinner, N.B. NOTCH2 Mutations Cause Alagille Syndrome, a Heterogeneous Disorder of the Notch Signaling Pathway. Am. J. Hum. Genet. 2006, 79, 169–173. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.J.; Kim, C.A.; Bertola, D.R.; Arantes, P.R.; Stewart, H.; Simpson, M.A.; Irving, M.D.; Robertson, S.P. Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu–Cheney syndrome. Eur. J. Hum. Genet. 2011, 20, 122–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldeman-Englert, C.R.; Jewett, T. 1q21.1 Recurrent Microdeletion. In Gene Reviews(R); Adam, M.P., Ardinger, H.H., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gamba, B.F.; Zechi-Ceide, R.M.; Kokitsu-Nakata, N.M.; Vendramini-Pittoli, S.; Rosenberg, C.; Santos, A.C.K.; Ribeiro-Bicudo, L.; Richieri-Costa, A. Interstitial 1q21.1 Microdeletion Is Associated with Severe Skeletal Anomalies, Dysmorphic Face and Moderate Intellectual Disability. Mol. Syndr. 2016, 7, 344–348. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.X.; Luo, G.N.; Wan, S.Q.; Qin, C.L.; Tang, J.; Zhang, M.; Du, M.; Xu, K.K.; Shi, J.Q. Detection of de novo genetic variants in Mayer-Rokitansky-Kuster-Hauser syndrome by whole genome sequencing. Eur. J. Obstet. Gynecol. Reprod. Biol. X 2019, 4, 100089. [Google Scholar] [CrossRef]
- Fisher, K.; Esham, R.H.; Thorneycroft, I. Scoliosis associated with typical Mayer-Rokitansky-Küster-Hauser syndrome. South. Med. J. 2000, 93, 243–246. [Google Scholar] [CrossRef]
- Chen, P.-C.J.; Yin, J.; Yu, H.-W.; Yuan, T.; Fernandez, M.; Yung, C.; Trinh, Q.M.; Peltekova, V.D.; Reid, J.G.; Tworog-Dube, E.; et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 11473–11478. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-K.; Chang, B.S.; Hong, Y.M.; Yang, S.W.; Seo, J.B. Spinal deformities in Noonan syndrome: A clinical review of sixty cases. J. Bone Jt. Surg. Am. Vol. 2001, 83, 1495–1502. [Google Scholar] [CrossRef]
- Li, P.; Yang, Y.M.; Sanchez, S.; Cui, D.C.; Dang, R.J.; Wang, X.Y.; Lin, Q.X.; Wang, Y.; Wang, C.; Chen, D.F.; et al. Deubiquitinase MYSM1 Is Essential for Normal Bone Formation and Mesenchymal Stem Cell Differentiation. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.-X.; Nguyen, Q.; Chou, Y.; Wang, T.; Nandakumar, V.; Yates, P.; Jones, L.; Wang, L.; Won, H.; Lee, H.-R.; et al. Control of B Cell Development by the Histone H2A Deubiquitinase MYSM1. Immunity 2011, 35, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Liakath-Ali, K.; Vancollie, V.; Heath, E.H.; Smedley, D.P.; Estabel, J.; Sunter, D.; DiTommaso, T.; White, J.K.; Ramirez-Solis, R.; Smyth, I.; et al. Novel skin phenotypes revealed by a genome-wide mouse reverse genetic screen. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiTommaso, T.; Jones, L.K.; Cottle, D.L.; Gerdin, A.-K.; Vancollie, V.E.; Watt, F.M.; Ramirez-Solis, R.; Bradley, A.; Steel, K.P.; Sundberg, J.P.; et al. Identification of Genes Important for Cutaneous Function Revealed by a Large Scale Reverse Genetic Screen in the Mouse. PLoS Genet. 2014, 10, e1004705. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, P.F.; Blank, R.D.; Raggio, C.L.; Merchant, S.; Jacobsen, F.S.; Faciszewski, T.; Shukla, S.K.; Greenlee, A.R.; Reynolds, C.; Schowalter, D.B. Congenital and Idiopathic Scoliosis: Clinical and Genetic Aspects. Clin. Med. Res. 2003, 1, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Wang, L.; Hu, J.; Zhao, S.; Liu, B.; Li, Y.; Du, H.; Zhang, Y.; Li, X.; Yan, Z.; et al. A Recurrent Rare SOX9 Variant (M469V) is Associated with Congenital Vertebral Malformations. Curr. Gene Ther. 2019, 19, 242–247. [Google Scholar] [CrossRef]
- Lin, M.; Zhao, S.; Liu, G.; Huang, Y.; Yu, C.; Zhao, Y.; Wang, L.; Zhang, Y.; Yan, Z.; Wang, S.; et al. Identification of novel FBN1 variations implicated in congenital scoliosis. J. Hum. Genet. 2019, 65, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Al Dhaheri, N.; Wu, N.; Zhao, S.; Wu, Z.; Blank, R.D.; Zhang, J.; Raggio, C.; Halanski, M.; Shen, J.; Noonan, K.; et al. KIAA1217: A novel candidate gene associated with isolated and syndromic vertebral malformations. Am. J. Med. Genet. Part A 2020, 182, 1664–1672. [Google Scholar] [CrossRef]
- Qian, F.; Germino, G.G. Mistakes happen: Somatic mutation and disease. Am. J. Hum. Genet. 1997, 61, 1000–1005. [Google Scholar] [CrossRef] [Green Version]
- Watnick, T.J.; Torres, V.E.; Gandolph, M.A.; Qian, F.; Onuchic, L.F.; Klinger, K.W.; Landes, G.; Germino, G.G. Somatic Mutation in Individual Liver Cysts Supports a Two-Hit Model of Cystogenesis in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell 1998, 2, 247–251. [Google Scholar] [CrossRef]
- Lamlum, H.; Ilyas, M.; Rowan, A.; Clark, S.; Johnson, V.; Bell, J.; Frayling, I.; Efstathiou, J.; Pack, K.; Payne, S.; et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: A new facet to Knudson’s ‘two-hit’ hypothesis. Nat. Med. 1999, 5, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Margraf, R.L.; VanSant-Webb, C.; Mao, R.; Viskochil, D.H.; Carey, J.; Hanson, H.; D’Astous, J.; Grossmann, A.; Stevenson, D.A. NF1 Somatic Mutation in Dystrophic Scoliosis. J. Mol. Neurosci. 2019, 68, 11–18. [Google Scholar] [CrossRef]

{kind=link}
| Gene | Patient | Type | Chr | Start | End | Size (bp) | Bayes Factor | Reads Ratio (Observed/ Expected) | Exons Annotation (hg19) (Gene_exon) | Inheritance Pattern | Highest Frequency in DGV (Sample Size >100) | gnomAD_ Structural_Variants Frequency (Heterozygous Loss) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NOTCH2 | CS033 | deletion | 1 | 120,611,949 | 120,612,020 | 71 | 4.62 | 0.657 | NOTCH2_1 | N.D. | 0.0037 | 0.00037 (0 in East Asia) |
| CS043 | deletion | 1 | 120,539,621 | 120,612,020 | 72,399 | 8.46 | 0.722 | NOTCH2_1-4 | De novo | 0.0037 | 0.00037 (0 in East Aisa) | |
| DSCAM | CS018 | deletion | 21 | 41,452,080 | 41,452,267 | 187 | 4.65 | 0.429 | DSCAM_25 | De novo | 0.00049 | N.A. |
| CS036 | deletion | 21 | 41,452,080 | 41,452,267 | 187 | 6.02 | 0.415 | DSCAM_25 | De novo | 0.00049 | N.A. | |
| CS050 | deletion | 21 | 41,452,080 | 41,452,267 | 187 | 5.8 | 0.468 | DSCAM_25 | Paternal | 0.00049 | N.A. | |
| CS053 | deletion | 21 | 41,452,080 | 41,452,267 | 187 | 5.45 | 0.494 | DSCAM_25 | N.D. | 0.00049 | N.A. | |
| CS064 | deletion | 21 | 41,452,080 | 41,452,267 | 187 | 6.1 | 0.463 | DSCAM_25 | N.D. | 0.00049 | N.A. | |
| SNTG1 | CS048 | deletion | 8 | 51,503,440 | 51,571,223 | 67,783 | 6.37 | 0.43 | SNTG1_13-15 | De novo | 0.0002 | 0.000046 (0 in East Asia) |
| TBX6 | CS059 | deletion | 16 | 29,674,601 | 30,199,897 | 525,296 | 644 | 0.555 | SPN_2,AC009133.19_2-3,QPRT_1-4,C16orf54_2,ZG16_2-4,KIF22_1-13,MAZ_1-5,PRRT2_2-3,PAGR1_1-3,CTD-2574D22.6_1-2,MVP_2-15,CDIPT_6-2,SEZ6L2_16-1,ASPHD1_1-3,KCTD13_6-1,TMEM219_1-4,TAOK2_2-16,HIRIP3_7-1,INO80E_1-7,DOC2A_11-2,C16orf92_2-3,FAM57B_5-1,ALDOA_8-16,PPP4C_2-9,TBX6_9-2,YPEL3_4-1,GDPD3_10-1,MAPK3_8-1,CORO1A_2-3,CORO1A_4-10 | N.D. | 0.0005 | 0.0001462 (0 in East Asia) |
| CS071 | deletion | 16 | 29,495,011 | 30,218,221 | 723,210 | 754 | 0.572 | NPIPL3_3-1,SPN_2,AC009133.19_2-3,QPRT_1-4,C16orf54_2,ZG16_3-4,KIF22_2-12,MAZ_1-5,PRRT2_2-3,PAGR1_1-3,CTD-2574D22.6_1-2,MVP_2-15,CDIPT_6-2,SEZ6L2_16-1,ASPHD1_1-3,KCTD13_6-1,TMEM219_1-4,TAOK2_2-16,HIRIP3_7-2,INO80E_1-7,DOC2A_11-2,C16orf92_2-3,FAM57B_5-1,ALDOA_8-16,PPP4C_2-9,TBX6_9-2,YPEL3_4-1,GDPD3_10-1,MAPK3_8-1,CORO1A_2-11,BOLA2B_3-1,SLX1A_1-5,SULT1A3_3-9,RP11-347C12.3_5-2 | De novo | 0.0005 | 0.0001462 (0 in East Asia) | |
| CS078 | deletion | 16 | 29,498,516 | 30,199,897 | 701,381 | 690 | 0.578 | NPIPL3_1,SPN_2,AC009133.19_2-3,QPRT_1-4,C16orf54_2,ZG16_3-4,KIF22_2-12,MAZ_1-5,PRRT2_2-3,PAGR1_1-3,CTD-2574D22.6_1-2, MVP_2-15,CDIPT_6-2,SEZ6L2_16-1,ASPHD1_1-3,KCTD13_6-1,TMEM219_1-4,TAOK2_2-16,HIRIP3_7-2,INO80E_1-7,DOC2A_11-2,C16orf92_2-3,FAM57B_5-1,ALDOA_8-16,PPP4C_2-9,TBX6_9-2,YPEL3_4-1,GDPD3_10-1,MAPK3_8-1,CORO1A_2-10 | N.D. | 0.0005 | 0.0001462 (0 in East Asia) | |
| CS081 | deletion | 16 | 29,498,516 | 30,199,897 | 701,381 | 645 | 0.558 | NPIPL3_1,SPN_2,AC009133.19_2-3,QPRT_1-4,C16orf54_2,ZG16_3-4,KIF22_2-12,MAZ_1-5,PRRT2_2-3,PAGR1_1-3,CTD-2574D22.6_1-2, MVP_2-15,CDIPT_6-2,SEZ6L2_16-1,ASPHD1_1-3,KCTD13_6-1,TMEM219_1-4,TAOK2_2-16,HIRIP3_7-2,INO80E_1-7,DOC2A_11-2,C16orf92_2-3,FAM57B_5-1,ALDOA_8-16,PPP4C_2-9,TBX6_9-2,YPEL3_4-1,GDPD3_10-1,MAPK3_8-1,CORO1A_2-10 | N.D. | 0.0005 | 0.0001462 (0 in East Asia) |
| Gene | Patient | Type | Chr | Start | End | Size (bp) | Bayes Factor | Reads Ratio (Observed/Expected) | Exons Annotation (hg19) (Gene_exon) | Inheritance Pattern | Highest Frequency in DGV (Sample Size > 100) | gnomAD_ Structural Variants Frequency (Heterozygous Loss) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NBPF20 | CS047 | deletion | 1 | 148,261,458 | 148,262,366 | 908 | 5.27 | 0.04 | NBPF20_98-99 | De novo | 0 | 0 (0 in East Asia) |
| FAM138C | CS048 | deletion | 9 | 35,061 | 35,519 | 458 | 6.51 | 0 | FAM138C_1-2 | * De novo | 0.0074 | N.A. |
| DHX40 | CS004 | deletion | 17 | 57,656,834 | 57,657,240 | 406 | 5.38 | 0 | DHX40_9-10 | N.D. | 0.0000922 | 0.0025 (0.008152 in East Asia) |
| CS035 | 4.91 | N.D. | ||||||||||
| CS043 | 6 | * De novo | ||||||||||
| CS050 | 7.23 | * De novo | ||||||||||
| CS053 | 7.02 | N.D. | ||||||||||
| CS057 | 6.29 | De novo |
| Gene | Type | Chr | Size (bp) | Count in 67 Patients | Count in 125 Controls | Highest Frequency in DGV (Sample Size > 100) | gnomad_ Structural_Variants Frequency (Heterozygous Loss) | gnomad_East Asia_Structural_Variants Frequency (Heterozygous Loss) |
|---|---|---|---|---|---|---|---|---|
| LRRC40 | deletion | 1 | 30,080–383,938 | 4 | 0 | 0.009556907 | 0.0000461 | 0.000 |
| SCN7A | deletion | 2 | 544–11,914 | 4 | 0 | 0.002257336 | 0.0000461 | 0.000 |
| MME | deletion | 3 | 279–55,232 | 4 | 0 | 0.000798722 | 0.0000462 | 0.000 |
| NAE1 | deletion | 16 | 6724–71,803 | 4 | 0 | N.A.b | 0 | 0.000 |
| TBX6 | deletion | 16 | 525,296–723,210 | 4 | 0 | 0.0005 | 0.0001462 | 0.000 |
| DHX40 | deletion | 17 | 107–2354 | 10 a | 1 | 0.0000922 | 0.0025 | 0.008152 |
| GMCL1 | deletion | 2 | 11,694–24,032 | 5 | 1 | 0.001303781 | 0.0000479 | 0.000 |
| MYSM1 | deletion | 1 | 190–10,053 | 4 | 1 | 0.0000922 | 0.0000461 | 0.0004139 |
| RASA2 | deletion | 3 | 2892–100,149 | 4 | 1 | 0.00086881 | 0 | 0.000 |
| NSMAF | deletion | 8 | 203–12,325 | 4 | 1 | 0.001145475 | 0 | 0.000 |
| MNS1 | deletion | 15 | 52,610–323,156 | 4 | 1 | 0.00518807 | 0.0000922 | 0.000 |
| PHKB | deletion | 16 | 7697–99,254 | 4 | 1 | 0.001198083 | 0.0000481 | 0.000 |
| SPO11 | deletion | 20 | 730–33,608 | 4 | 1 | 0.00064226 | 0 | 0.000 |
| ABCA6 | duplication | 17 | 560–13,580 | 4 | 1 | 0.0009219 | 0 | 0.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, W.; Feng, X.; Yue, M.; Cheung, P.W.H.; Choi, V.N.T.; Song, Y.-Q.; Luk, K.D.K.; Cheung, J.P.Y.; Gao, B. Identification of Copy Number Variants in a Southern Chinese Cohort of Patients with Congenital Scoliosis. Genes 2021, 12, 1213. https://doi.org/10.3390/genes12081213
Lai W, Feng X, Yue M, Cheung PWH, Choi VNT, Song Y-Q, Luk KDK, Cheung JPY, Gao B. Identification of Copy Number Variants in a Southern Chinese Cohort of Patients with Congenital Scoliosis. Genes. 2021; 12(8):1213. https://doi.org/10.3390/genes12081213
Chicago/Turabian StyleLai, Wenjing, Xin Feng, Ming Yue, Prudence W. H. Cheung, Vanessa N. T. Choi, You-Qiang Song, Keith D. K. Luk, Jason Pui Yin Cheung, and Bo Gao. 2021. "Identification of Copy Number Variants in a Southern Chinese Cohort of Patients with Congenital Scoliosis" Genes 12, no. 8: 1213. https://doi.org/10.3390/genes12081213
APA StyleLai, W., Feng, X., Yue, M., Cheung, P. W. H., Choi, V. N. T., Song, Y.-Q., Luk, K. D. K., Cheung, J. P. Y., & Gao, B. (2021). Identification of Copy Number Variants in a Southern Chinese Cohort of Patients with Congenital Scoliosis. Genes, 12(8), 1213. https://doi.org/10.3390/genes12081213