
Severity of Idiopathic Scoliosis Is Associated with Differential Methylation: An Epigenome-Wide Association Study of Monozygotic Twins with Idiopathic Scoliosis

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. DNA Methylation Processing
2.3. Statistical Analysis
2.4. Prioritization of Candidates: Blood Methylation Correlation
3. Results
3.1. Demographics
3.2. Discordant Curvature Analysis
3.3. Curve Severity Analysis
3.4. Candidate Prioritization
4. Discussions
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cases | Control | ||||||
|---|---|---|---|---|---|---|---|
| Median | Min | Max | Median | Min | Max | p Value | |
| CD8T Cell (%) | 11.5% | 8.3% | 33.6% | 12.2% | 6.9% | 18.7% | 0.1953 |
| CD4T Cell (%) | 18.5% | 7.7% | 28.3% | 14.3% | 9.3% | 18.0% | 0.1953 |
| Natural Killer Cell Count (%) | 5.3% | 2.2% | 8.8% | 5.7% | 1.9% | 8.4% | 0.7422 |
| Bcell Count (%) | 5.8% | 3.5% | 10.3% | 6.2% | 2.9% | 10.3% | 0.6406 |
| Monocyte Cell Count (%) | 7.5% | 3.4% | 9.5% | 8.0% | 5.1% | 10.9% | 0.6406 |
| Neutrophil Cell Count (%) | 55.3% | 25.7% | 67.2% | 58.4% | 52.0% | 72.9% | 0.1953 |
| Feature/Gene | Chr. | Start Position | End Position | Number of Probes | Nominal p Value | FDR p Value | Maximum Bone Correlation | FDR p Value for Maximum Bone Correlation | Percent Strongly Positively Correlated Probes Across DMR |
|---|---|---|---|---|---|---|---|---|---|
| WNT10A | chr2 | 219,744,145 | 219,745,748 | 9 | 2.17 × 10−5 | 0.0113 | 0.83 | 0.0307 | 33.3% |
| BCL2L2 * | chr14 | 23,775,206 | 23,776,530 | 8 | 2.17 × 10−5 | 0.0113 | 0.50 | 0.4069 | 14.3% |
| CRISP2 | chr6 | 49,681,178 | 49,681,774 | 11 | 2.19 × 10−5 | 0.0113 | 0.89 | 0.0128 | 100.0% |
| SLFN13 | chr17 | 33,773,921 | 33,777,219 | 11 | 2.19 × 10−5 | 0.0113 | −0.27 | 0.7380 | 0.0% |
| RBPJL | chr20 | 43,934,854 | 43,935,551 | 12 | 2.20 × 10−5 | 0.0113 | 0.72 | 0.1048 | 66.7% |
| KDM2B | chr12 | 122,018,574 | 122,020,205 | 14 | 2.21 × 10−5 | 0.0113 | 0.83 | 0.0336 | 50.0% |
| MS4A3 | chr11 | 59,822,727 | 59,828,426 | 13 | 2.21 × 10−5 | 0.0113 | −0.57 | 0.2917 | 7.7% |
| GPR21 | chr9 | 125,794,756 | 125,797,284 | 14 | 2.21 × 10−5 | 0.0113 | 0.38 | 0.5940 | 0.0% |
| IL27 | chr16 | 28,518,114 | 28,519,597 | 9 | 4.34 × 10−5 | 0.0156 | 0.75 | 0.0844 | 33.3% |
| AZU1 | chr19 | 826,359 | 827,821 | 9 | 4.34 × 10−5 | 0.0156 | −0.28 | 0.7263 | 0.0% |
| CA14 | chr1 | 150,229,143 | 150,230,345 | 9 | 6.51 × 10−5 | 0.0196 | 0.78 | 0.0585 | 33.3% |
| HTRA4 | chr8 | 38,830,814 | 38,831,857 | 11 | 6.58 × 10−5 | 0.0196 | 0.29 | 0.7117 | 0.0% |
| ESRP2 | chr16 | 68,269,763 | 68,271,177 | 13 | 6.62 × 10−5 | 0.0196 | 0.35 | 0.6385 | 0.0% |
| ELANE | chr19 | 850,975 | 852,311 | 8 | 8.68 × 10−5 | 0.0199 | 0.45 | 0.4788 | 0.0% |
| CLDN15 | chr7 | 100,880,751 | 100,882,286 | 10 | 8.74 × 10−5 | 0.0199 | 0.68 | 0.1542 | 20.0% |
| RUNX1 | chr21 | 36,259,179 | 36,422,112 | 14 | 8.85 × 10−5 | 0.0199 | 0.60 | 0.2518 | 21.4% |
| NFE2 | chr12 | 54,689,278 | 54,696,210 | 14 | 8.85 × 10−5 | 0.0199 | −0.36 | 0.6204 | 0.0% |
| C6orf229 | chr6 | 24,799,059 | 24,799,757 | 5 | 9.45 × 10−5 | 0.0199 | 0.70 | 0.1293 | 20.0% |
| SLC25A16 | chr10 | 70,287,181 | 70,287,493 | 6 | 1.07 × 10−5 | 0.0216 | −0.45 | 0.4813 | 0.0% |
| EPHA2 | chr1 | 16,482,553 | 16,483,528 | 8 | 1.09 × 10−4 | 0.0216 | 0.47 | 0.4470 | 0.0% |
| TNFSF13 | chr17 | 7,460,690 | 7,462,249 | 12 | 1.32 × 10−4 | 0.0223 | 0.65 | 0.1888 | 8.3% |
| UBASH3A | chr21 | 43,822,540 | 43,823,863 | 6 | 1.69 × 10−4 | 0.0252 | −0.64 | 0.1916 | 0.0% |
| C7orf49 | chr7 | 134,852,662 | 134,855,381 | 9 | 1.74 × 10−4 | 0.0256 | 0.59 | 0.2703 | 11.1% |
| ADAP2 | chr17 | 29,247,612 | 29,248,848 | 9 | 1.95 × 10−4 | 0.0266 | −0.39 | 0.5675 | 0.0% |
| AMT | chr3 | 49,459,855 | 49,461,563 | 11 | 1.97 × 10−4 | 0.0266 | 0.37 | 0.5997 | 0.0% |
| C9orf47 | chr9 | 91,604,473 | 91,606,140 | 12 | 2.64 × 10−4 | 0.0318 | 0.79 | 0.0518 | 18.2% |
| CD3D | chr11 | 118,213,272 | 118,214,927 | 8 | 2.78 × 10−4 | 0.0318 | −0.20 | 0.8262 | 0.0% |
| HSPB6 | chr19 | 36,247,867 | 36,248,907 | 8 | 2.82 × 10−4 | 0.0318 | 0.24 | 0.7811 | 0.0% |
| STAB1 | chr3 | 52,528,714 | 52,529,393 | 8 | 3.04 × 10−4 | 0.0329 | 0.79 | 0.0534 | 12.5% |
| ACY3 | chr11 | 67,415,183 | 67,418,365 | 8 | 3.26 × 10−4 | 0.0329 | 0.72 | 0.1045 | 62.5% |
| MPG | chr16 | 125,896 | 128,009 | 11 | 3.29 × 10−4 | 0.0329 | 0.82 | 0.0368 | 10.0% |
| KLRD1 | chr12 | 10,455,788 | 10,460,639 | 8 | 3.34 × 10−4 | 0.0329 | −0.73 | 0.1030 | 12.5% |
| FES | chr15 | 91,427,184 | 91,428,456 | 10 | 3.50 × 10−4 | 0.033 | 0.39 | 0.5737 | 0.0% |
| ESM1 | chr5 | 54,281,198 | 54,282,459 | 13 | 3.97 × 10−4 | 0.036 | 0.79 | 0.0526 | 61.5% |
| CTSG | chr14 | 25,045,625 | 25,046,267 | 6 | 4.28 × 10−4 | 0.0382 | −0.36 | 0.6151 | 0.0% |
| HMGN2 | chr1 | 26,797,576 | 267,987,40 | 6 | 4.71 × 10−4 | 0.0403 | −0.45 | 0.4827 | 0.0% |
| LRG1 | chr19 | 4,540,003 | 4,540,782 | 6 | 4.71 × 10−4 | 0.0403 | −0.42 | 0.5212 | 0.0% |
| LOC100130933 | chr17 | 73,641,809 | 73,642,991 | 10 | 4.81 × 10−4 | 0.0405 | 0.44 | 0.5036 | 0.0% |
| HMGCR | chr5 | 74,632,477 | 74,637,028 | 5 | 5.10 × 10−4 | 0.0419 | 0.71 | 0.1165 | 20.0% |
| RPSAP52 | chr12 | 66,220,754 | 66,221,950 | 6 | 5.14 × 10−4 | 0.0419 | −0.77 | 0.0683 | 0.0% |
| MIR145 | chr5 | 148,808,721 | 148,810,180 | 7 | 5.18 × 10−4 | 0.0419 | −0.38 | 0.5867 | 0.0% |
| PILRA | chr7 | 99,970,448 | 99,971,016 | 5 | 5.31 × 10−4 | 0.0419 | 0.37 | 0.6031 | 0.0% |
| TMEM219 | chr16 | 29,972,752 | 29,974,294 | 6 | 5.57 × 10−4 | 0.0431 | 0.77 | 0.0667 | 66.7% |
| GIMAP1 | chr7 | 150,412,503 | 150,415,143 | 5 | 5.86 × 10−4 | 0.0451 | 0.33 | 0.6550 | 0.0% |
| CREBBP | chr16 | 3,930,112 | 3,931,489 | 5 | 6.16 × 10−4 | 0.0457 | 0.76 | 0.0725 | 80.0% |
| MAST2 | chr1 | 46,268,158 | 46,269,120 | 6 | 6.21 × 10−4 | 0.0457 | 0.70 | 0.1248 | 16.7% |
| TAGLN3 | chr3 | 111,717,534 | 111,718,245 | 8 | 6.29 × 10−4 | 0.0457 | 0.68 | 0.1531 | 12.5% |
| S100P | chr4 | 6,694,923 | 6,695,698 | 9 | 6.30 × 10−4 | 0.0457 | −0.51 | 0.3861 | 0.0% |
| FAM53C | chr5 | 137,672,901 | 137,675,418 | 7 | 6.48 × 10−4 | 0.0458 | −0.25 | 0.7705 | 0.0% |
| GAMT | chr19 | 1,401,372 | 1,402,626 | 5 | 6.58 × 10−4 | 0.0459 | −0.35 | 0.6314 | 0.0% |
| H6PD | chr1 | 9,293,583 | 9,303,499 | 10 | 6.77 × 10−4 | 0.0462 | 0.39 | 0.5714 | 0.0% |
| ITGAE | chr17 | 3,704,471 | 37,058,75 | 10 | 6.77 × 10−4 | 0.0462 | −0.45 | 0.4790 | 0.0% |
| CDH9 | chr5 | 27,036,352 | 27,040,099 | 10 | 6.77 × 10−4 | 0.0462 | −0.42 | 0.5366 | 0.0% |
| ELP2 | chr18 | 33,709,151 | 33,709,799 | 7 | 6.91 × 10−4 | 0.0462 | −0.22 | 0.8067 | 0.0% |
| LRP11 | chr6 | 150,185,188 | 150,186,488 | 8 | 7.16 × 10−4 | 0.0462 | 0.54 | 0.3423 | 12.5% |
| FIGLA | chr2 | 71,017,541 | 71,018,823 | 11 | 7.46 × 10−4 | 0.0473 | 0.61 | 0.2324 | 18.2% |
| CAPN13 | chr2 | 31,020,802 | 31,031,755 | 6 | 8.07 × 10−4 | 0.0496 | 0.48 | 0.4419 | 0.0% |
| Category | Fold Enrichment | Bonferroni Adjusted p Value |
|---|---|---|
| Gene Ontology Cellular Component | ||
| secretory granule lumen (GO:0034774) | 3.8 | 7.29 × 10−3 |
| Feature/Gen | Chr. | Start Position | End Position | Number of Probes | Nominal p Value | FDR p Value | Maximum Bone Correlation | FDR p Value for Maximum Bone Correlation | Percent Strongly Positively Correlated Probes Across DMR |
|---|---|---|---|---|---|---|---|---|---|
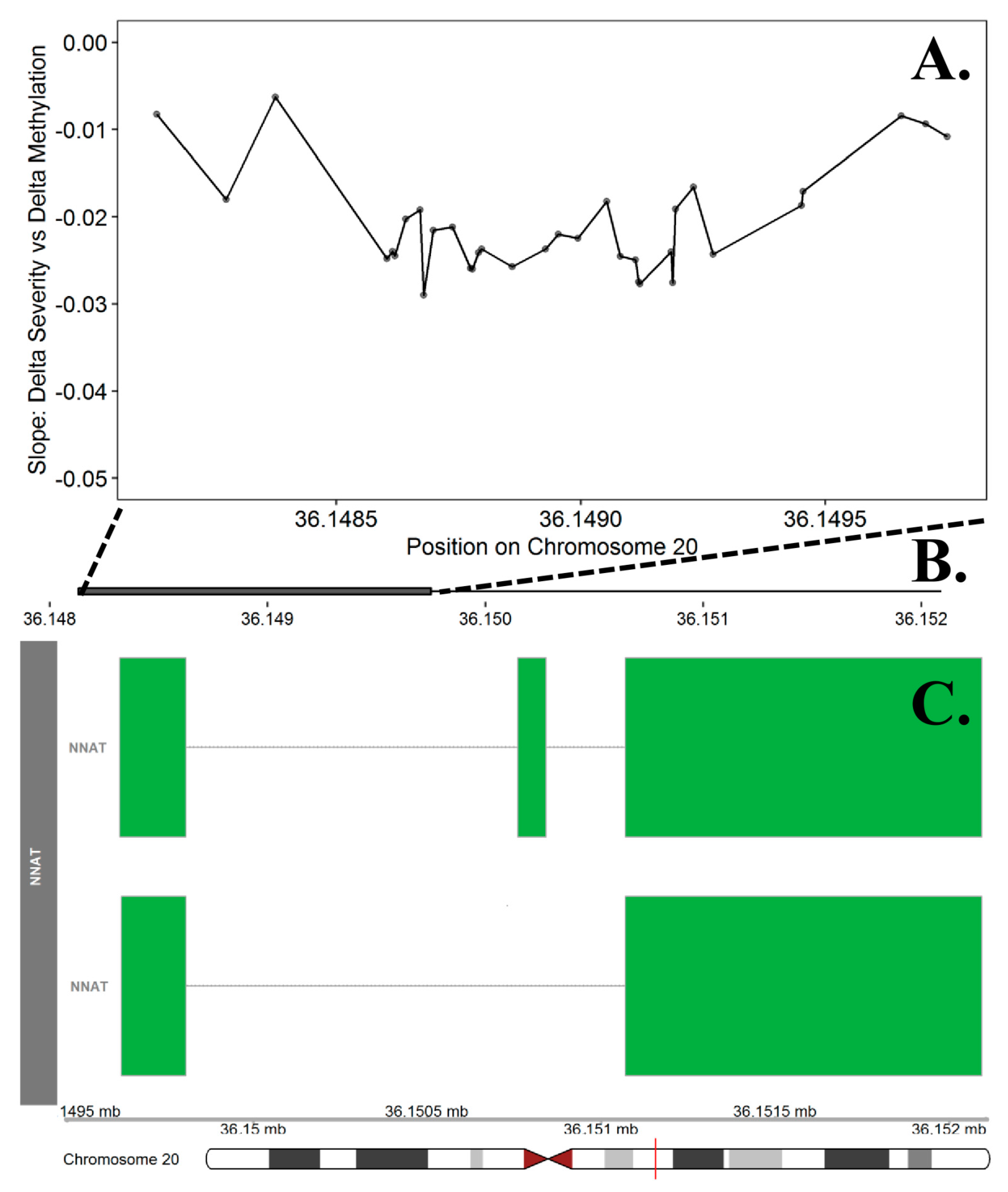
| NNAT | chr20 | 36,148,133 | 36,149,750 | 34 | 1.11 × 10−5 | 0.0237 | 0.57 | 0.2898 | 12.1% |
| TMEM232 | chr5 | 110,021,543 | 110,062,837 | 11 | 5.09 × 10−5 | 0.0237 | 0.68 | 0.1441 | 30.0% |
| SLC22A20 | chr11 | 64,979,837 | 64,981,596 | 9 | 8.43 × 10−5 | 0.0252 | 0.66 | 0.1680 | 33.3% |
| PDE12 | chr3 | 57,541,377 | 57,543,243 | 5 | 9.89 × 10−5 | 0.0252 | 0.69 | 0.1347 | 40.0% |
| SUV420H2 | chr19 | 55,850,082 | 55,852,507 | 5 | 1.98 × 10−4 | 0.0299 | 0.70 | 0.1324 | 40.0% |
| CYR61 | chr1 | 86,045,347 | 86,046,661 | 10 | 2.22 × 10−4 | 0.0299 | 0.71 | 0.1148 | 40.0% |
| ZNF440 | chr19 | 11,924,860 | 11,925,219 | 7 | 2.63 × 10−4 | 0.0333 | 0.63 | 0.2110 | 14.3% |
| LOC150622 | chr2 | 6,072,139 | 6,072,801 | 5 | 3.16 × 10−4 | 0.0348 | 0.61 | 0.2383 | 40.0% |
| GANC | chr15 | 42,565,522 | 42,566,390 | 7 | 3.38 × 10−4 | 0.0357 | 0.88 | 0.0153 | 28.6% |
| TMEM87A | chr15 | 42,565,522 | 42,566,390 | 7 | 3.38 × 10−4 | 0.0357 | NA | NA | NA |
| NME3 | chr16 | 1,821,559 | 1,822,346 | 8 | 3.59 × 10−4 | 0.0366 | 0.76 | 0.0729 | 37.5% |
| SLC6A5 | chr11 | 20,619,598 | 20,621,109 | 8 | 3.59 × 10−4 | 0.0366 | 0.80 | 0.0489 | 28.6% |
| HSPB6 | chr19 | 36,247,867 | 36,248,907 | 8 | 3.98 × 10−4 | 0.0378 | 0.24 | 0.7811 | 0.0% |
| RAB22A | chr20 | 56,883,532 | 56,885,003 | 8 | 4.38 × 10−4 | 0.0391 | 0.78 | 0.0594 | 25.0% |
| SLC1A1 | chr9 | 4,489,544 | 4,490,288 | 6 | 4.60 × 10−4 | 0.0394 | 0.60 | 0.2551 | 16.7% |
| ACTN4 | chr19 | 39,137,911 | 39,138,334 | 7 | 4.89 × 10−4 | 0.0407 | 0.84 | 0.0298 | 33.3% |
| PRKD1 | chr14 | 30,396,845 | 30,397,763 | 6 | 4.96 × 10−4 | 0.0407 | 0.49 | 0.4133 | 16.7% |
| STL | chr6 | 125,284,212 | 125,284,659 | 6 | 4.96 × 10−4 | 0.0407 | 0.22 | 0.8030 | 0.0% |
| CPXM1 | chr20 | 2,781,122 | 2,782,348 | 9 | 5.06 × 10−4 | 0.0411 | 0.43 | 0.5202 | 0.0% |
| INSR | chr19 | 7,294,087 | 7,295,192 | 5 | 5.27 × 10−4 | 0.0413 | 0.61 | 0.2438 | 20.0% |
| NPY | chr7 | 24,322,873 | 24,324,570 | 8 | 5.58 × 10−4 | 0.0421 | 0.84 | 0.0276 | 37.5% |
| RAB38 | chr11 | 87,908,558 | 87,909,729 | 9 | 6.33 × 10−4 | 0.045 | 0.73 | 0.0995 | 44.4% |
| CNTNAP5 | chr2 | 124,782,117 | 124,783,254 | 9 | 6.33 × 10−4 | 0.045 | 0.63 | 0.2146 | 11.1% |
| AKR7L | chr1 | 19,600,471 | 19,601,069 | 7 | 7.52 × 10−4 | 0.0495 | 0.49 | 0.4219 | 0.0% |
| COPB1 | chr11 | 14,521,639 | 14,522,617 | 6 | 7.79 × 10−4 | 0.0495 | 0.88 | 0.0149 | 50.0% |
| MEI1 | chr22 | 42,095,347 | 42,095,536 | 5 | 7.91 × 10−4 | 0.0495 | 0.65 | 0.1891 | 40.0% |
| PPP2R1B | chr11 | 111,637,044 | 111,638,422 | 5 | 7.91 × 10−4 | 0.0495 | 0.69 | 0.1381 | 40.0% |
| KCNB1 | chr20 | 48,098,642 | 48,100,238 | 8 | 7.97 × 10−4 | 0.0495 | 0.55 | 0.3315 | 12.5% |
| Category | Fold Enrichment | Bonferroni Adjusted p Value |
|---|---|---|
| Gene Ontology Molecular Function | ||
| RNA polymerase II cis-regulatory region sequence-specific DNA binding (GO:0000978) | 2.4 | 1.27 × 10−3 |
| DNA-binding transcription factor activity, RNA polymerase II-specific (GO:0000981) | 2.3 | 3.65 × 10−4 |
| regulatory region nucleic acid binding (GO:0001067) | 2.2 | 1.04 × 10−3 |
| transcription regulator activity (GO:0140110) | 1.9 | 2.04 × 10−2 |
| Gene Ontology Cellular Component | ||
| chromatin (GO:0000785) | 2.3 | 7.60 × 10−3 |
| Gene Ontology Biologic Process | ||
| pituitary gland development (GO:0021983) | 12.0 | 8.09 × 10−3 |
| anterior/posterior pattern specification (GO:0009952) | 5.2 | 3.91 × 10−3 |
| mesenchyme development (GO:0060485) | 4.8 | 8.84 × 10−3 |
| heart morphogenesis (GO:0003007) | 4.4 | 2.44 × 10−3 |
| embryonic organ development (GO:0048568) | 3.3 | 4.70 × 10−3 |
| negative regulation of cell differentiation (GO:0045596) | 3.0 | 6.46 × 10−3 |
| head development (GO:0060322) | 2.7 | 2.37 × 10−3 |
| tube development (GO:0035295) | 2.7 | 3.04 × 10−3 |
| neuron differentiation (GO:0030182) | 2.5 | 5.09 × 10−3 |
| anatomical structure morphogenesis (GO:0009653) | 2.0 | 3.30 × 10−3 |
References
- Altaf, F.; Drinkwater, J.; Phan, K.; Cree, A.K. Systematic Review of School Scoliosis Screening. Spine Deform. 2017, 5, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.L.; Dolan, L.A.; Cheng, J.C.; Danielsson, A.; Morcuende, J.A. Adolescent idiopathic scoliosis. Lancet 2008, 371, 1527–1537. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.H. Genetics of familial idiopathic scoliosis. Clin. Orthop. Relat. Res. 2007, 462, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Khanshour, A.M.; Wise, C.A. The Genetic Architecture of Adolescent Idiopathic Scoliosis. In Pathogenesis of Idiopathic Scoliosis; Springer: Berlin, Germany, 2018; pp. 51–74. [Google Scholar]
- Tang, N.L.; Yeung, H.Y.; Hung, V.W.; Di Liao, C.; Lam, T.P.; Yeung, H.M.; Lee, K.M.; Ng, B.K.; Cheng, J.C. Genetic epidemiology and heritability of AIS: A study of 415 Chinese female patients. J. Orthop Res. 2012, 30, 1464–1469. [Google Scholar] [CrossRef]
- Ward, K.; Ogilvie, J.; Argyle, V.; Nelson, L.; Meade, M.; Braun, J.; Chettier, R. Polygenic inheritance of adolescent idiopathic scoliosis: A study of extended families in Utah. Am. J. Med. Genet. A 2010, 152, 1178–1188. [Google Scholar] [CrossRef]
- Miller, N.H.; Mims, B.; Child, A.; Milewicz, D.M.; Sponseller, P.; Blanton, S.H. Genetic analysis of structural elastic fiber and collagen genes in familial adolescent idiopathic scoliosis. J. Orthop. Res. 1996, 14, 994–999. [Google Scholar] [CrossRef]
- Miller, N.H.; Schwab, D.L.; Sponseller, P.; Shugert, E.; Bell, J.; Maestri, N. Genomic Search for X-Linkage in Familial Adolescent Idiopathic Scoliosis. In Research into Spinal Deformities 2; Stokes, I.A., Ed.; IOS Press: Amsterdam, The Netherlands, 1998; pp. 209–213. [Google Scholar]
- Wise, C.A.; Barnes, R.; Gillum, J.; Herring, J.A.; Bowcock, A.M.; Lovett, M. Localization of susceptibility to familial idiopathic scoliosis. Spine (Phila Pa 1976) 2000, 25, 2372–2380. [Google Scholar] [CrossRef]
- Chan, V.; Fong, G.C.; Luk, K.D.; Yip, B.; Lee, M.K.; Wong, M.S.; Lu, D.D.; Chan, T.K. A genetic locus for adolescent idiopathic scoliosis linked to chromosome 19p13.3. Am. J. Hum. Genet. 2002, 71, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Justice, C.M.; Miller, N.H.; Marosy, B.; Zhang, J.; Wilson, A.F. Familial idiopathic scoliosis: Evidence of an X-linked susceptibility locus. Spine (Phila Pa 1976) 2003, 28, 589–594. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.H.; Justice, C.M.; Marosy, B.; Doheny, K.F.; Pugh, E.; Zhang, J.; Dietz, H.C., 3rd; Wilson, A.F. Identification of candidate regions for familial idiopathic scoliosis. Spine (Phila Pa 1976) 2005, 30, 1181–1187. [Google Scholar] [CrossRef]
- Alden, K.J.; Marosy, B.; Nzegwu, N.; Justice, C.M.; Wilson, A.F.; Miller, N.H. Idiopathic scoliosis: Identification of candidate regions on chromosome 19p13. Spine (Phila Pa 1976) 2006, 31, 1815–1819. [Google Scholar] [CrossRef]
- Miller, N.H.; Marosy, B.; Justice, C.M.; Novak, S.M.; Tang, E.Y.; Boyce, P.; Pettengil, J.; Doheny, K.F.; Pugh, E.W.; Wilson, A.F. Linkage analysis of genetic loci for kyphoscoliosis on chromosomes 5p13, 13q13.3, and 13q32. Am. J. Med. Genet. A 2006, 140, 1059–1068. [Google Scholar] [CrossRef]
- Montanaro, L.; Parisini, P.; Greggi, T.; Di Silvestre, M.; Campoccia, D.; Rizzi, S.; Arciola, C.R. Evidence of a linkage between matrilin-1 gene (MATN1) and idiopathic scoliosis. Scoliosis 2006, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Gurnett, C.A.; Alaee, F.; Bowcock, A.; Kruse, L.; Lenke, L.G.; Bridwell, K.H.; Kuklo, T.; Luhmann, S.J.; Dobbs, M.B. Genetic linkage localizes an adolescent idiopathic scoliosis and pectus excavatum gene to chromosome 18 q. Spine (Phila Pa 1976) 2009, 34, E94–E100. [Google Scholar] [CrossRef] [Green Version]
- Marosy, B.; Justice, C.M.; Vu, C.; Zorn, A.; Nzegwu, N.; Wilson, A.F.; Miller, N.H. Identification of susceptibility loci for scoliosis in FIS families with triple curves. Am. J. Med. Genet. A 2010, 152, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.G.; Alvarado, D.M.; Haller, G.E.; Cruchaga, C.; Harms, M.B.; Zhang, T.; Willing, M.C.; Grange, D.K.; Braverman, A.C.; Miller, N.H.; et al. Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum. Mol. Genet. 2014, 23, 5271–5282. [Google Scholar] [CrossRef] [Green Version]
- Baschal, E.E.; Wethey, C.I.; Swindle, K.; Baschal, R.M.; Gowan, K.; Tang, N.L.; Alvarado, D.M.; Haller, G.E.; Dobbs, M.B.; Taylor, M.R.; et al. Exome sequencing identifies a rare HSPG2 variant associated with familial idiopathic scoliosis. G3 2014, 5, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Patten, S.A.; Margaritte-Jeannin, P.; Bernard, J.C.; Alix, E.; Labalme, A.; Besson, A.; Girard, S.L.; Fendri, K.; Fraisse, N.; Biot, B.; et al. Functional variants of POC5 identified in patients with idiopathic scoliosis. J. Clin. Investig. 2015, 125, 1124–1128. [Google Scholar] [CrossRef]
- Grauers, A.; Wang, J.; Einarsdottir, E.; Simony, A.; Danielsson, A.; Akesson, K.; Ohlin, A.; Halldin, K.; Grabowski, P.; Tenne, M.; et al. Candidate gene analysis and exome sequencing confirm LBX1 as a susceptibility gene for idiopathic scoliosis. Spine J. 2015, 15, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, Y.; Zhang, L.; Guo, H.; Tian, D.; Li, Y.; Peng, Y.; Zheng, Y.; Dai, Y.; Xia, K.; et al. AKAP2 identified as a novel gene mutated in a Chinese family with adolescent idiopathic scoliosis. J. Med. Genet. 2016, 53, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Haller, G.; Alvarado, D.; McCall, K.; Yang, P.; Cruchaga, C.; Harms, M.; Goate, A.; Willing, M.; Morcuende, J.A.; Baschal, E.; et al. A polygenic burden of rare variants across extracellular matrix genes among individuals with adolescent idiopathic scoliosis. Hum. Mol. Genet. 2016, 25, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Chen, C.; Zhou, T.; Yang, S.; Gao, B.; Zhou, H.; Lian, C.; Wu, Z.; Qiu, X.; Yang, X.; et al. Rare coding variants in MAPK7 predispose to adolescent idiopathic scoliosis. Hum. Mutat. 2017, 38, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Einarsdottir, E.; Grauers, A.; Wang, J.; Jiao, H.; Escher, S.A.; Danielsson, A.; Simony, A.; Andersen, M.; Christensen, S.B.; Akesson, K.; et al. CELSR2 is a candidate susceptibility gene in idiopathic scoliosis. PLoS ONE 2017, 12, e0189591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baschal, E.E.; Terhune, E.A.; Wethey, C.I.; Baschal, R.M.; Robinson, K.D.; Cuevas, M.T.; Pradhan, S.; Sutphin, B.S.; Taylor, M.R.G.; Gowan, K.; et al. Idiopathic Scoliosis Families Highlight Actin-Based and Microtubule-Based Cellular Projections and Extracellular Matrix in Disease Etiology. G3 2018, 8, 2663–2672. [Google Scholar] [CrossRef] [Green Version]
- Terhune, E.A.; Cuevas, M.T.; Monley, A.M.; Wethey, C.I.; Chen, X.; Cattell, M.V.; Bayrak, M.N.; Bland, M.R.; Sutphin, B.; Trahan, G.D.; et al. Mutations in KIF7 implicated in idiopathic scoliosis in humans and axial curvatures in zebrafish. Hum. Mutat. 2021, 42, 392–407. [Google Scholar] [CrossRef]
- Sharma, S.; Gao, X.; Londono, D.; Devroy, S.E.; Mauldin, K.N.; Frankel, J.T.; Brandon, J.M.; Zhang, D.; Li, Q.Z.; Dobbs, M.B.; et al. Genome-wide association studies of adolescent idiopathic scoliosis suggest candidate susceptibility genes. Hum. Mol. Genet. 2011, 20, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kou, I.; Takahashi, A.; Johnson, T.A.; Kono, K.; Kawakami, N.; Uno, K.; Ito, M.; Minami, S.; Yanagida, H.; et al. A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet. 2011, 43, 1237–1240. [Google Scholar] [CrossRef]
- Nelson, L.M.; Chettier, R.; Ogilvie, J.W.; Ward, K. Candidate Genes for Susceptibility of Adolescent Idiopathic Scoliosis Identified Through a Large Genome-Wide Association Study. In Proceedings of the Scoliosis Research Society 46th Annual Meeting & Course, Louisville, KY, USA, 14–17 September 2011. [Google Scholar]
- Kou, I.; Takahashi, Y.; Johnson, T.A.; Takahashi, A.; Guo, L.; Dai, J.; Qiu, X.; Sharma, S.; Takimoto, A.; Ogura, Y.; et al. Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat. Genet. 2013, 45, 676–679. [Google Scholar] [CrossRef]
- Miyake, A.; Kou, I.; Takahashi, Y.; Johnson, T.A.; Ogura, Y.; Dai, J.; Qiu, X.; Takahashi, A.; Jiang, H.; Yan, H.; et al. Identification of a susceptibility locus for severe adolescent idiopathic scoliosis on chromosome 17q24.3. PLoS ONE 2013, 8, e72802. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Tang, N.L.; Xu, L.; Qin, X.; Mao, S.; Song, Y.; Liu, L.; Li, F.; Liu, P.; Yi, L.; et al. Genome-wide association study identifies new susceptibility loci for adolescent idiopathic scoliosis in Chinese girls. Nat. Commun. 2015, 6, 8355. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Kou, I.; Miura, S.; Takahashi, A.; Xu, L.; Takeda, K.; Takahashi, Y.; Kono, K.; Kawakami, N.; Uno, K.; et al. A Functional SNP in BNC2 Is Associated with Adolescent Idiopathic Scoliosis. Am. J. Hum. Genet. 2015, 97, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Londono, D.; Eckalbar, W.L.; Gao, X.; Zhang, D.; Mauldin, K.; Kou, I.; Takahashi, A.; Matsumoto, M.; Kamiya, N.; et al. A PAX1 enhancer locus is associated with susceptibility to idiopathic scoliosis in females. Nat. Commun. 2015, 6, 6452. [Google Scholar] [CrossRef] [Green Version]
- Kou, I.; Otomo, N.; Takeda, K.; Momozawa, Y.; Lu, H.F.; Kubo, M.; Kamatani, Y.; Ogura, Y.; Takahashi, Y.; Nakajima, M.; et al. Genome-wide association study identifies 14 previously unreported susceptibility loci for adolescent idiopathic scoliosis in Japanese. Nat. Commun. 2019, 10, 3685. [Google Scholar] [CrossRef]
- Mogha, A.; Benesh, A.E.; Patra, C.; Engel, F.B.; Schoneberg, T.; Liebscher, I.; Monk, K.R. Gpr126 functions in Schwann cells to control differentiation and myelination via G-protein activation. J. Neurosci. 2013, 33, 17976–17985. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.F.; Yang, G.H.; Pan, X.H.; Zhang, S.J.; Zhao, C.; Qiu, B.S.; Gu, H.F.; Hong, J.F.; Cao, L.; Chen, Y.; et al. Association of GPR126 gene polymorphism with adolescent idiopathic scoliosis in Chinese populations. Genomics 2015, 105, 101–107. [Google Scholar] [CrossRef]
- Karner, C.M.; Long, F.; Solnica-Krezel, L.; Monk, K.R.; Gray, R.S. Gpr126/Adgrg6 deletion in cartilage models idiopathic scoliosis and pectus excavatum in mice. Hum. Mol. Genet. 2015, 24, 4365–4373. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Xu, L.; Xia, C.; Zhu, W.; Sun, W.; Liu, Z.; Qiu, Y.; Zhu, Z. Genetic Variant of GPR126 Gene is Functionally Associated With Adolescent Idiopathic Scoliosis in Chinese Population. Spine (Phila Pa 1976) 2017, 42, E1098–E1103. [Google Scholar] [CrossRef]
- Man, G.C.; Tang, N.L.; Chan, T.F.; Lam, T.P.; Li, J.W.; Ng, B.K.; Zhu, Z.; Qiu, Y.; Cheng, J.C. Replication Study for the Association of GWAS-associated Loci with Adolescent Idiopathic Scoliosis Susceptibility and Curve Progression in a Chinese Population. Spine (Phila Pa 1976) 2019, 44, 464–471. [Google Scholar] [CrossRef]
- Kou, I.; Watanabe, K.; Takahashi, Y.; Momozawa, Y.; Khanshour, A.; Grauers, A.; Zhou, H.; Liu, G.; Fan, Y.H.; Takeda, K.; et al. A multi-ethnic meta-analysis confirms the association of rs6570507 with adolescent idiopathic scoliosis. Sci. Rep. 2018, 8, 11575. [Google Scholar] [CrossRef]
- Liu, G.; Liu, S.; Lin, M.; Li, X.; Chen, W.; Zuo, Y.; Liu, J.; Niu, Y.; Zhao, S.; Long, B.; et al. Genetic polymorphisms of GPR126 are functionally associated with PUMC classifications of adolescent idiopathic scoliosis in a Northern Han population. J. Cell Mol. Med. 2018, 22, 1964–1971. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Peng, Y.; Liang, G.; Liang, A.; Ye, W.; Zhang, L.; Sharma, S.; Su, P.; Huang, D. Association between common variants near LBX1 and adolescent idiopathic scoliosis replicated in the Chinese Han population. PLoS ONE 2013, 8, e53234. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Yang, Q.; Liu, Y.; Guan, Y.; Zhan, X.; Xiao, Z.; Wei, Q. Association between ladybird homeobox 1 gene polymorphisms and adolescent idiopathic scoliosis: A MOOSE-compliant meta-analysis. Medicine (Baltimore) 2019, 98, e16314. [Google Scholar] [CrossRef]
- Liang, J.; Xing, D.; Li, Z.; Chua, S.; Li, S. Association Between rs11190870 Polymorphism Near LBX1 and Susceptibility to Adolescent Idiopathic Scoliosis in East Asian Population: A Genetic Meta-Analysis. Spine (Phila Pa 1976) 2014, 39, 862–869. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, L.; Roffey, D.M.; Phan, P.; Wai, E.K. Association of rs11190870 near LBX1 with adolescent idiopathic scoliosis in East Asians: A systematic review and meta-analysis. Spine J. 2014, 14, 2968–2975. [Google Scholar] [CrossRef]
- Jiang, H.; Qiu, X.; Dai, J.; Yan, H.; Zhu, Z.; Qian, B.; Qiu, Y. Association of rs11190870 near LBX1 with adolescent idiopathic scoliosis susceptibility in a Han Chinese population. Eur. Spine J. 2013, 22, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.L.; Gao, S.J.; Xu, H.; Liu, Y.; Li, H.L.; Chen, X.Y.; Ning, G.Z.; Feng, S.Q. The association of rs11190870 near LBX1 with the susceptibility and severity of AIS, a meta-analysis. Int. J. Surg. 2018, 54, 193–200. [Google Scholar] [CrossRef]
- Cao, Y.; Min, J.; Zhang, Q.; Li, H.; Li, H. Associations of LBX1 gene and adolescent idiopathic scoliosis susceptibility: A meta-analysis based on 34,626 subjects. BMC Musculoskelet Disord 2016, 17, 309. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Yamashita, H.; Kou, I.; Takimoto, A.; Meguro-Horike, M.; Horike, S.; Sakuma, T.; Miura, S.; Adachi, T.; Yamamoto, T.; et al. Functional Investigation of a Non-coding Variant Associated with Adolescent Idiopathic Scoliosis in Zebrafish: Elevated Expression of the Ladybird Homeobox Gene Causes Body Axis Deformation. PLoS Genet. 2016, 12, e1005802. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wu, N.; Zuo, Y.; Zhou, Y.; Liu, J.; Liu, Z.; Chen, W.; Liu, G.; Chen, Y.; Chen, J.; et al. Genetic Polymorphism of LBX1 Is Associated with Adolescent Idiopathic Scoliosis in Northern Chinese Han Population. Spine (Phila Pa 1976) 2017, 42, 1125–1129. [Google Scholar] [CrossRef]
- Xu, L.; Wu, Z.; Xia, C.; Tang, N.; Cheng, J.C.Y.; Qiu, Y.; Zhu, Z. A Genetic Predictive Model Estimating the Risk of Developing Adolescent Idiopathic Scoliosis. Curr. Genom. 2019, 20, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Chettier, R.; Nelson, L.; Ogilvie, J.W.; Albertsen, H.M.; Ward, K. Haplotypes at LBX1 have distinct inheritance patterns with opposite effects in adolescent idiopathic scoliosis. PLoS ONE 2015, 10, e0117708. [Google Scholar] [CrossRef]
- Londono, D.; Kou, I.; Johnson, T.A.; Sharma, S.; Ogura, Y.; Tsunoda, T.; Takahashi, A.; Matsumoto, M.; Herring, J.A.; Lam, T.P.; et al. A meta-analysis identifies adolescent idiopathic scoliosis association with LBX1 locus in multiple ethnic groups. J. Med. Genet. 2014, 51, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Nada, D.; Julien, C.; Samuels, M.E.; Moreau, A. A Replication Study for Association of LBX1 Locus with Adolescent Idiopathic Scoliosis in French-Canadian Population. Spine (Phila Pa 1976) 2018, 43, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.H.; Song, Y.Q.; Chan, D.; Takahashi, Y.; Ikegawa, S.; Matsumoto, M.; Kou, I.; Cheah, K.S.; Sham, P.; Cheung, K.M.; et al. SNP rs11190870 near LBX1 is associated with adolescent idiopathic scoliosis in southern Chinese. J. Hum. Genet. 2012, 57, 244–246. [Google Scholar] [CrossRef] [Green Version]
- Edery, P.; Margaritte-Jeannin, P.; Biot, B.; Labalme, A.; Bernard, J.C.; Chastang, J.; Kassai, B.; Plais, M.H.; Moldovan, F.; Clerget-Darpoux, F. New disease gene location and high genetic heterogeneity in idiopathic scoliosis. Eur. J. Hum. Genet. 2011, 19, 865–869. [Google Scholar] [CrossRef]
- Gorman, K.F.; Julien, C.; Moreau, A. The genetic epidemiology of idiopathic scoliosis. Eur. Spine J. 2012, 21, 1905–1919. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.G.; Edgar, M.; Margulies, J.Y.; Miller, N.H.; Raso, V.J.; Reinker, K.A.; Rivard, C.H. Etiology of idiopathic scoliosis: Current trends in research. J. Bone Jt. Surg. Am. 2000, 82, 1157–1168. [Google Scholar] [CrossRef]
- Dunwoodie, S.L.; Kusumi, K. The Genetics and Development of Scoliosis, 2nd ed.; Springer International Publishing: Cham, Switzerland, 2018; p. 1. [Google Scholar] [CrossRef]
- Heary, R.F.; Madhavan, K. Genetics of scoliosis. Neurosurgery 2008, 63, 222–227. [Google Scholar] [CrossRef]
- Kruse, L.M.; Buchan, J.G.; Gurnett, C.A.; Dobbs, M.B. Polygenic threshold model with sex dimorphism in adolescent idiopathic scoliosis: The Carter effect. J. Bone Jt. Surg. Am. 2012, 94, 1485–1491. [Google Scholar] [CrossRef] [Green Version]
- Arpon, A.; Milagro, F.I.; Ramos-Lopez, O.; Mansego, M.L.; Riezu-Boj, J.I.; Martinez, J.A.; Project, M. Methylome-Wide Association Study in Peripheral White Blood Cells Focusing on Central Obesity and Inflammation. Genes 2019, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Grauers, A.; Rahman, I.; Gerdhem, P. Heritability of scoliosis. Eur. Spine J. 2012, 21, 1069–1074. [Google Scholar] [CrossRef] [Green Version]
- Domcke, S.; Bardet, A.F.; Adrian Ginno, P.; Hartl, D.; Burger, L.; Schubeler, D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 2015, 528, 575–579. [Google Scholar] [CrossRef]
- Gutierrez-Arcelus, M.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Ongen, H.; Yurovsky, A.; Bryois, J.; Giger, T.; Romano, L.; Planchon, A.; et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife 2013, 2, e00523. [Google Scholar] [CrossRef]
- Lappalainen, T.; Greally, J.M. Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 2017, 18, 441–451. [Google Scholar] [CrossRef]
- Gertz, J.; Varley, K.E.; Reddy, T.E.; Bowling, K.M.; Pauli, F.; Parker, S.L.; Kucera, K.S.; Willard, H.F.; Myers, R.M. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011, 7, e1002228. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Fernandez, A.F.; Sainz, J.; Zarrabeitia, M.T.; Sanudo, C.; Garcia-Renedo, R.; Perez-Nunez, M.I.; Garcia-Ibarbia, C.; Fraga, M.F.; Riancho, J.A. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 2013, 65, 197–205. [Google Scholar] [CrossRef]
- Mohandas, N.; Bass-Stringer, S.; Maksimovic, J.; Crompton, K.; Loke, Y.J.; Walstab, J.; Reid, S.M.; Amor, D.J.; Reddihough, D.; Craig, J.M. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin. Epigenet. 2018, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Diboun, I.; Wani, S.; Ralston, S.H.; Albagha, O.M. Epigenetic analysis of Paget’s disease of bone identifies differentially methylated loci that predict disease status. eLife 2021, 10, e65715. [Google Scholar] [CrossRef]
- Mao, S.H.; Qian, B.P.; Shi, B.; Zhu, Z.Z.; Qiu, Y. Quantitative evaluation of the relationship between COMP promoter methylation and the susceptibility and curve progression of adolescent idiopathic scoliosis. Eur. Spine J. 2018, 27, 272–277. [Google Scholar] [CrossRef]
- Shi, B.; Xu, L.; Mao, S.; Xu, L.; Liu, Z.; Sun, X.; Zhu, Z.; Qiu, Y. Abnormal PITX1 gene methylation in adolescent idiopathic scoliosis: A pilot study. BMC Musculoskelet Disord 2018, 19, 138. [Google Scholar] [CrossRef]
- Janusz, P.; Chmielewska, M.; Andrusiewicz, M.; Kotwicka, M.; Kotwicki, T. Methylation of Estrogen Receptor 1 Gene in the Paraspinal Muscles of Girls with Idiopathic Scoliosis and Its Association with Disease Severity. Genes 2021, 12, 790. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, M.; Janusz, P.; Andrusiewicz, M.; Kotwicki, T.; Kotwicka, M. Methylation of estrogen receptor 2 (ESR2) in deep paravertebral muscles and its association with idiopathic scoliosis. Sci. Rep. 2020, 10, 22331. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, L.; Wang, X.; Yan, Z.; Yang, X.; Lin, M.; Liu, S.; Zuo, Y.; Niu, Y.; Zhao, S.; et al. Whole-Genome Methylation Analysis of Phenotype Discordant Monozygotic Twins Reveals Novel Epigenetic Perturbation Contributing to the Pathogenesis of Adolescent Idiopathic Scoliosis. Front. Bioeng. Biotechnol. 2019, 7, 364. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Lin, T.; Liang, S.; Gao, R.; Jiang, H.; Shao, W.; Yang, F.; Zhou, X. Value of DNA methylation in predicting curve progression in patients with adolescent idiopathic scoliosis. EBioMedicine 2018, 36, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Shands, A.R.J.; Eisberg, H.B. The incidence of scoliosis in the state of Delaware; a study of 50,000 minifilms of the chest made during a survey for tuberculosis. J. Bone Jt. Surg. Am. 1955, 37, 1243–1249. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Logue, M.W.; Smith, A.K.; Wolf, E.J.; Maniates, H.; Stone, A.; Schichman, S.A.; McGlinchey, R.E.; Milberg, W.; Miller, M.W. The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples. Epigenomics 2017, 9, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Irizarry, R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genom. Biol. 2014, 15, R31. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Martorell-Marugan, J.; Gonzalez-Rumayor, V.; Carmona-Saez, P. mCSEA: Detecting subtle differentially methylated regions. Bioinformatics 2019, 35, 3257–3262. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Mi, H.; Thomas, P. PANTHER pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, P.; Luthman, H.; McGuigan, F.E.; Akesson, K.E. Epigenome-wide cross-tissue correlation of human bone and blood DNA methylation—can blood be used as a surrogate for bone? Epigenetics 2020, 16, 92–105. [Google Scholar] [CrossRef]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7, 13555. [Google Scholar] [CrossRef]
- Webster, A.P.; Plant, D.; Ecker, S.; Zufferey, F.; Bell, J.T.; Feber, A.; Paul, D.S.; Beck, S.; Barton, A.; Williams, F.M.K.; et al. Increased DNA methylation variability in rheumatoid arthritis-discordant monozygotic twins. Genom. Med. 2018, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Takebayashi, H.; Bepari, A.K.; Esumi, S.; Yanagawa, Y.; Tamamaki, N. Dpy19l1, a multi-transmembrane protein, regulates the radial migration of glutamatergic neurons in the developing cerebral cortex. Development 2011, 138, 4979–4990. [Google Scholar] [CrossRef]
- Watanabe, K.; Bizen, N.; Sato, N.; Takebayashi, H. Endoplasmic Reticulum-Localized Transmembrane Protein Dpy19L1 Is Required for Neurite Outgrowth. PLoS ONE 2016, 11, e0167985. [Google Scholar] [CrossRef] [Green Version]
- Data, Z.H. Phenotype Annotation. 1994–2006. Available online: https://zfin.org/ZDB-FIG-070117-942 (accessed on 17 May 2021).
- Perry, R.B.; Hezroni, H.; Goldrich, M.J.; Ulitsky, I. Regulation of Neuroregeneration by Long Noncoding RNAs. Mol. Cell 2018, 72, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, X.; Shen, J.; Zhang, L.; Chan, M.T.V.; Wu, W.K.K. Emerging roles of non-coding RNAs in scoliosis. Cell Prolif. 2020, 53, e12736. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Sirbu, I.O.; Duester, G. Retinoic-acid signalling in node ectoderm and posterior neural plate directs left-right patterning of somitic mesoderm. Nat. Cell Biol. 2006, 8, 271–277. [Google Scholar] [CrossRef]
- Hitier, M.; Hamon, M.; Denise, P.; Lacoudre, J.; Thenint, M.A.; Mallet, J.F.; Moreau, S.; Quarck, G. Lateral Semicircular Canal Asymmetry in Idiopathic Scoliosis: An Early Link between Biomechanical, Hormonal and Neurosensory Theories? PLoS ONE 2015, 10, e0131120. [Google Scholar] [CrossRef]
- Rousie, D.; Joly, O.; Berthoz, A. Posterior Basicranium asymmetry and idiopathic scoliosis. arXiv 2009, arXiv:0911.5042. [Google Scholar]
- Carry, P.M.; Duke, V.R.; Brazell, C.J.; Stence, N.; Scholes, M.; Rousie, D.L.; Hadley Miller, N. Lateral semi-circular canal asymmetry in females with idiopathic scoliosis. PLoS ONE 2020, 15, e0232417. [Google Scholar] [CrossRef]
- Hino, H.; Araki, K.; Uyama, E.; Takeya, M.; Araki, M.; Yoshinobu, K.; Miike, K.; Kawazoe, Y.; Maeda, Y.; Uchino, M.; et al. Myopathy phenotype in transgenic mice expressing mutated PABPN1 as a model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2004, 13, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.M. Neuronatin gene: Imprinted and misfolded: Studies in Lafora disease, diabetes and cancer may implicate NNAT-aggregates as a common downstream participant in neuronal loss. Genomics 2014, 103, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikyo, N.; Williamson, C.M.; John, R.M.; Barton, S.C.; Beechey, C.V.; Ball, S.T.; Cattanach, B.M.; Surani, M.A.; Peters, J. Genetic and functional analysis of neuronatin in mice with maternal or paternal duplication of distal Chr 2. Dev. Biol. 1997, 190, 66–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, H.K.; Wylie, A.A.; Murphy, S.K.; Jirtle, R.L. The neuronatin gene resides in a “micro-imprinted” domain on human chromosome 20q11. 2. Genomics 2001, 77, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.J.; Herzog, H. NPY regulation of bone remodelling. Neuropeptides 2009, 43, 457–463. [Google Scholar] [CrossRef]
- Wu, J.Q.; Jiang, N.; Yu, B. Mechanisms of action of neuropeptide Y on stem cells and its potential applications in orthopaedic disorders. World J. Stem. Cells 2020, 12, 986–1000. [Google Scholar] [CrossRef]
- Xu, M.; Horrell, J.; Snitow, M.; Cui, J.; Gochnauer, H.; Syrett, C.M.; Kallish, S.; Seykora, J.T.; Liu, F.; Gaillard, D.; et al. WNT10A mutation causes ectodermal dysplasia by impairing progenitor cell proliferation and KLF4-mediated differentiation. Nat. Commun. 2017, 8, 15397. [Google Scholar] [CrossRef] [Green Version]
- Vink, C.P.; Ockeloen, C.W.; ten Kate, S.; Koolen, D.A.; Ploos van Amstel, J.K.; Kuijpers-Jagtman, A.M.; van Heumen, C.C.; Kleefstra, T.; Carels, C.E. Variability in dentofacial phenotypes in four families with WNT10A mutations. Eur. J. Hum. Genet. 2014, 22, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Cawthorn, W.P.; Bree, A.J.; Yao, Y.; Du, B.; Hemati, N.; Martinez-Santibanez, G.; MacDougald, O.A. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 2012, 50, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Grimes, D.T.; Boswell, C.W.; Morante, N.F.; Henkelman, R.M.; Burdine, R.D.; Ciruna, B. Zebrafish models of idiopathic scoliosis link cerebrospinal fluid flow defects to spine curvature. Science 2016, 352, 1341–1344. [Google Scholar] [CrossRef] [Green Version]
- Gurnett, C.A.; Alaee, F.; Kruse, L.M.; Desruisseau, D.M.; Hecht, J.T.; Wise, C.A.; Bowcock, A.M.; Dobbs, M.B. Asymmetric lower-limb malformations in individuals with homeobox PITX1 gene mutation. Am. J. Hum. Genet. 2008, 83, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef]
- Hamilton, J.P. Epigenetics: Principles and practice. Dig. Dis. 2011, 29, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
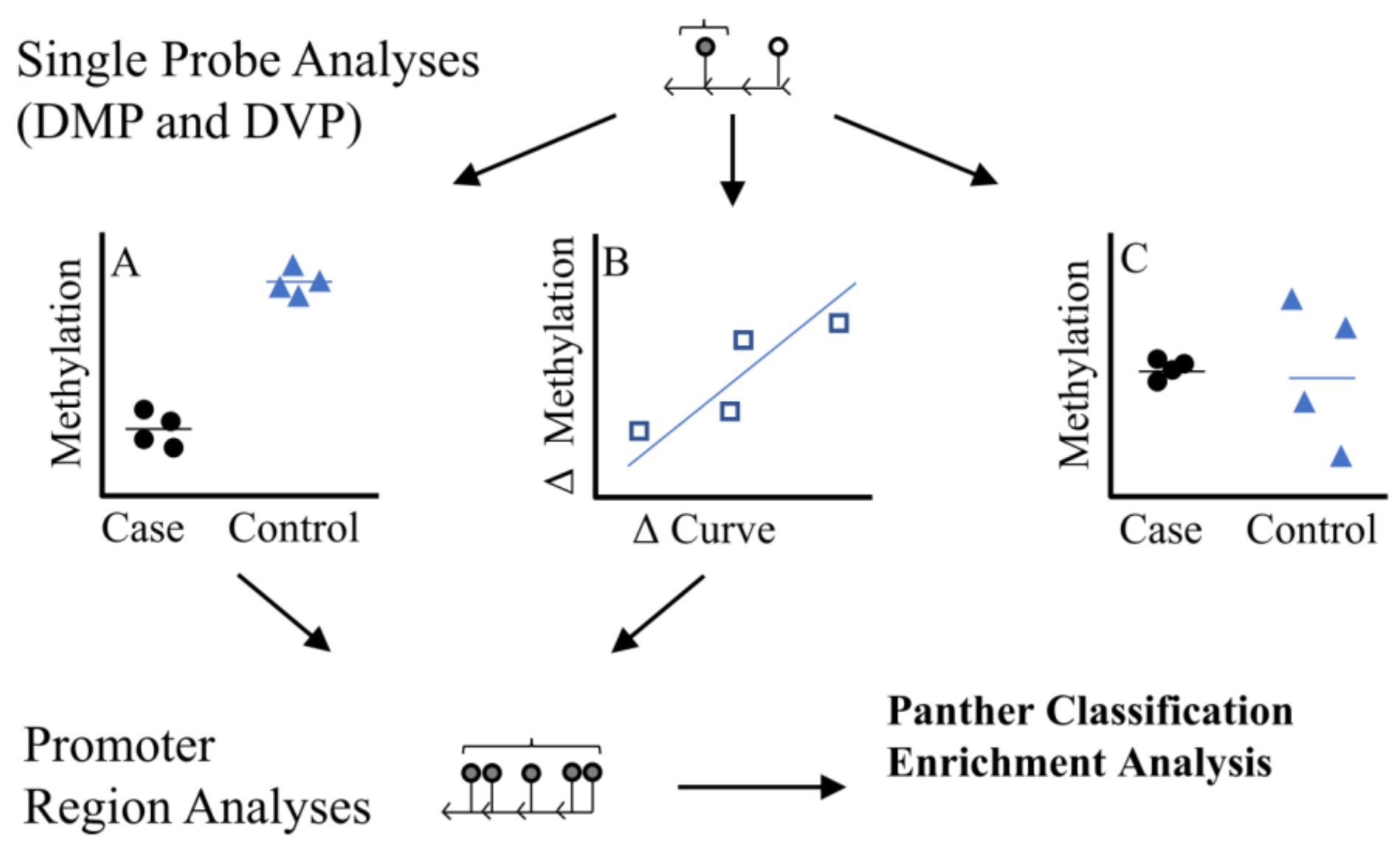
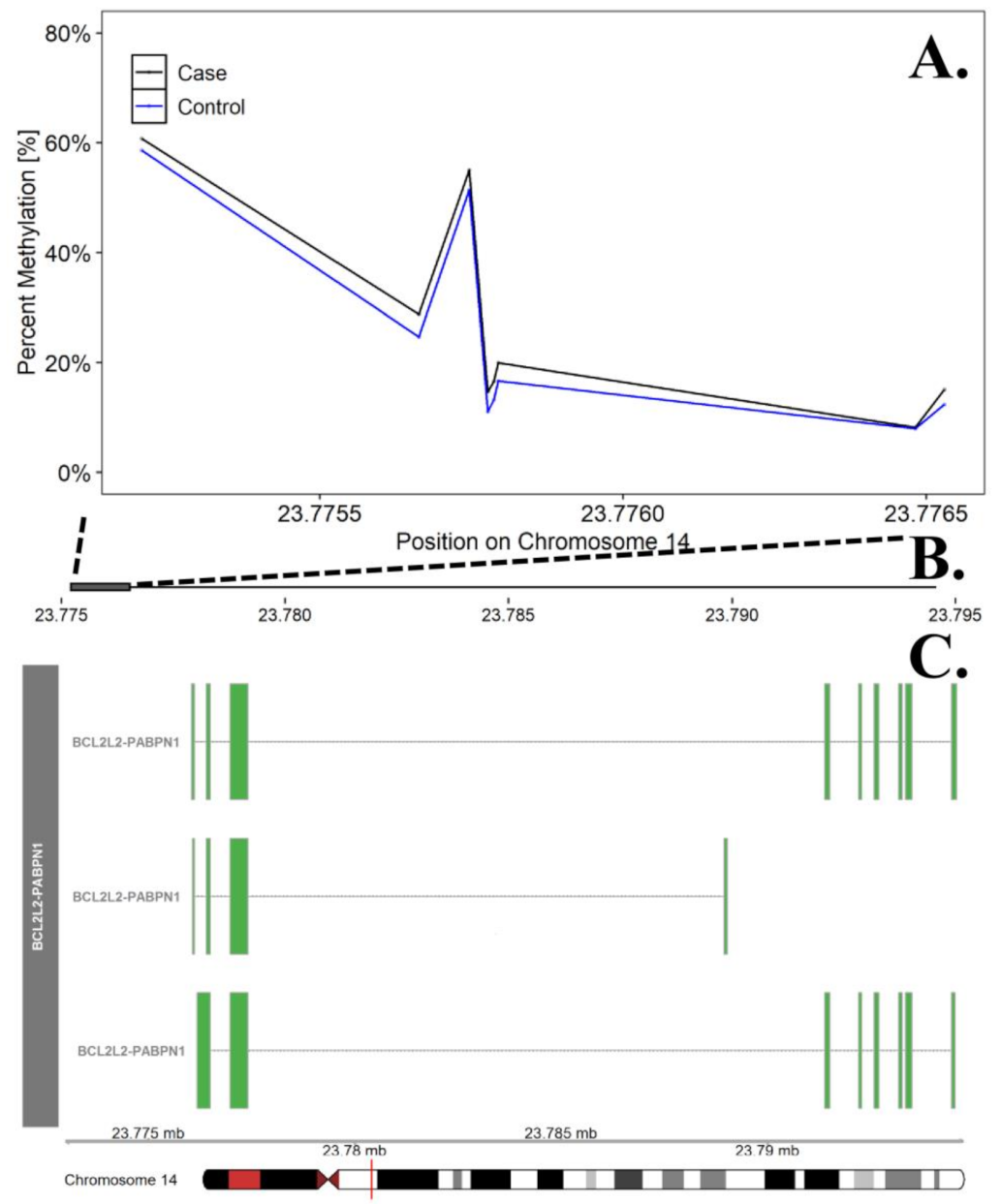
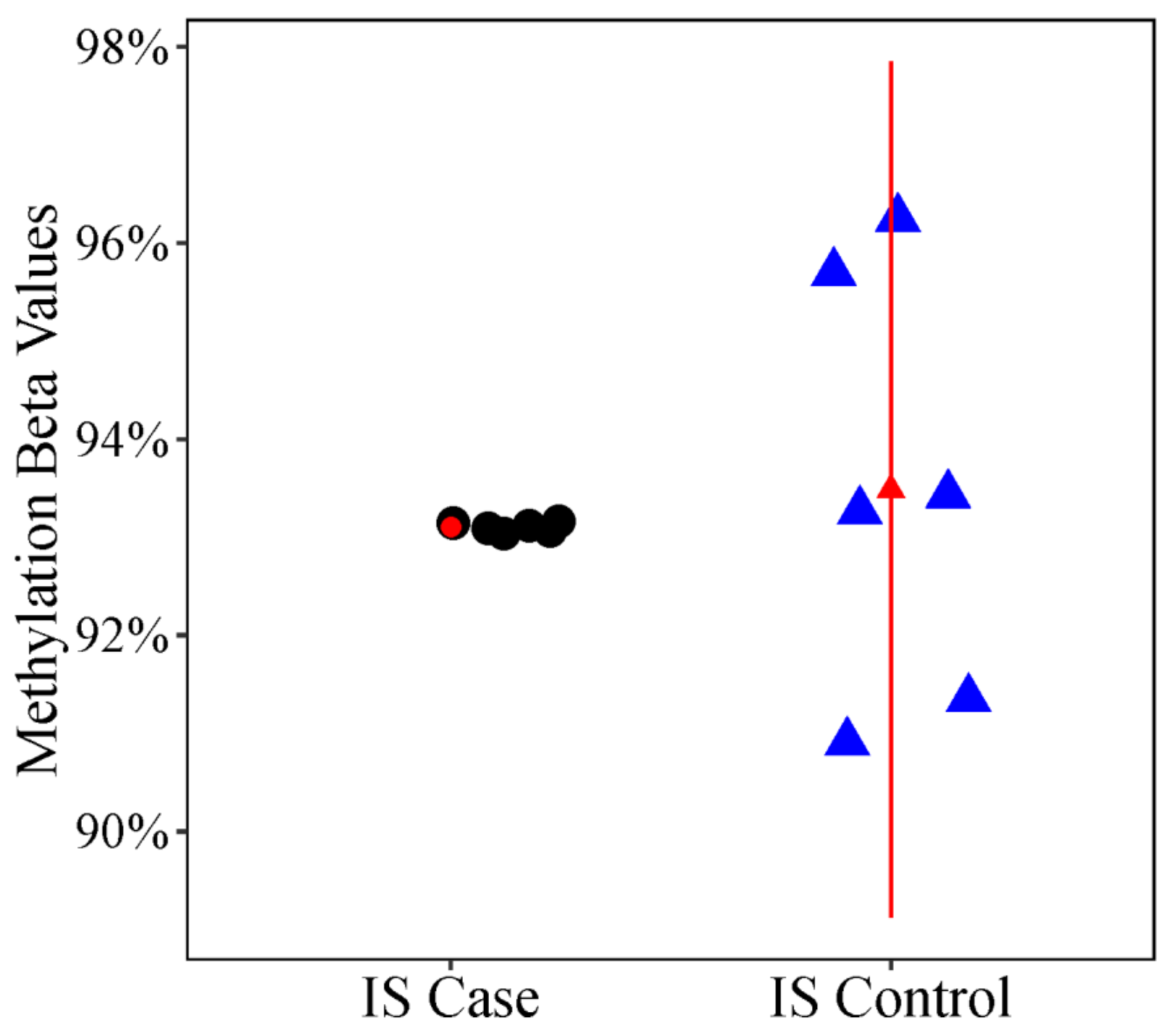
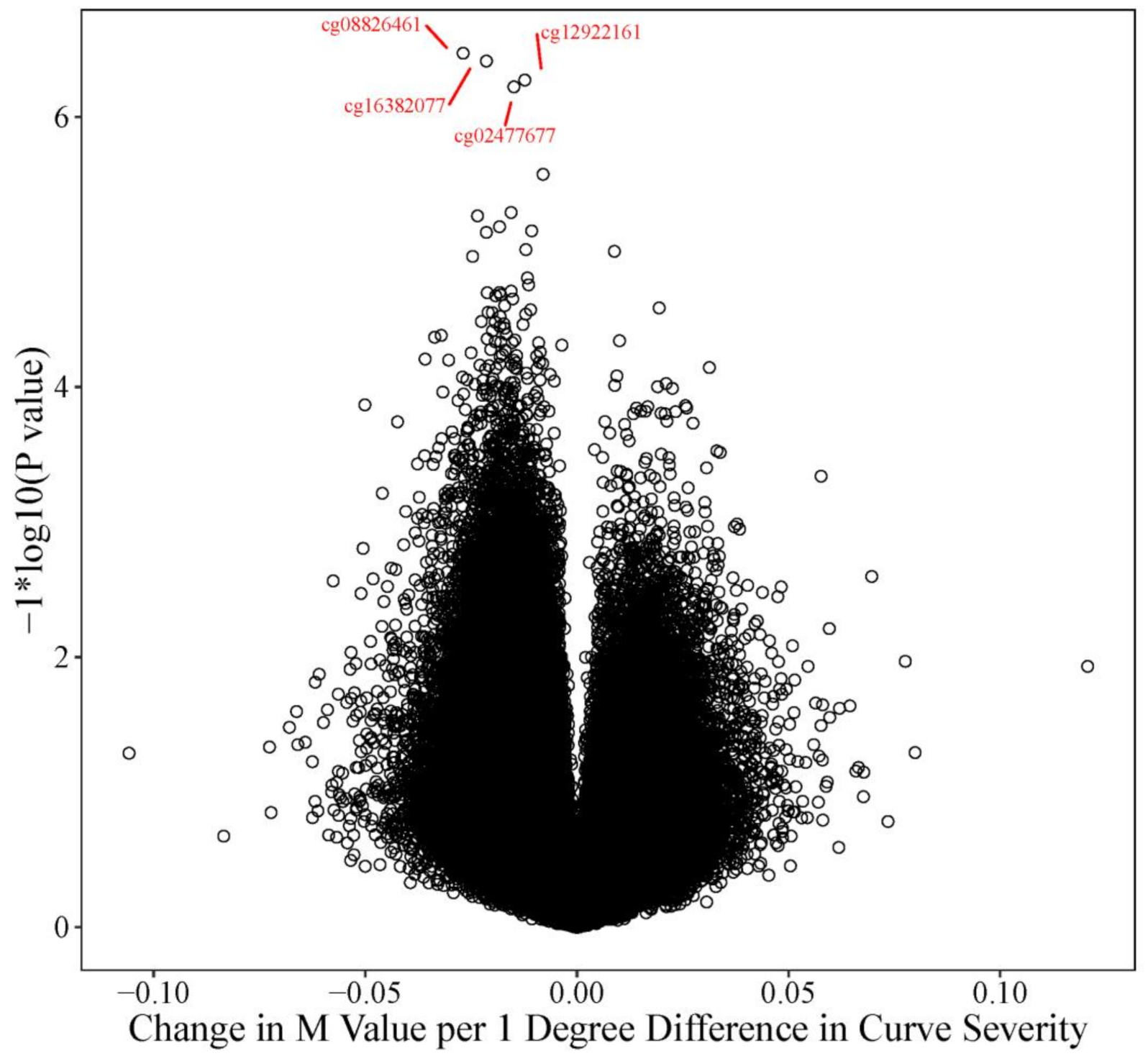
| Twin Pair | ID | Case Status | Curve Degree † | Age * |
|---|---|---|---|---|
| Discordant | ||||
| 1 | 15505 | Case | 48 | 44.8 |
| 15501 | Control | 41/37 | 44.8 | |
| 2 | 15643 | Case | 75 | 81.4 |
| 15642 | Control | 35 | 81.9 | |
| 3 | 16012 | Case | 50 | 16.3 |
| 16009 | Control | 22 | 16.3 | |
| 4 | 16615 | Case | 52/48 | 33.4 |
| 16611 | Control | 32/28 | 33.3 | |
| 5 | 18453 | Case | 34 | 5.6 |
| 18454 | Control | 23 | 5.6 | |
| 6 | 19294 | Case | 56/43 | 48.7 |
| 19292 | Control | 12 | 48.7 | |
| Concordant | ||||
| 7 | 16037 | NA | 45 | 42.8 |
| 16038 | NA | 45 | 42.8 | |
| 8 | 18721 | NA | 26/33 | 25.3 |
| 18722 | NA | 29/31 | 25.5 | |
| Nearest Gene | Chr. | Start Position | End Position | Number of Probes | DMR Nominal p Value | DMR FDR p Value | Maximum Bone Correlation | FDR Adj. p Value for Maximum Bone Correlation | Percent Strongly Positively Correlated Probes Across DMR |
|---|---|---|---|---|---|---|---|---|---|
| Discordant DMR Analysis | |||||||||
| WNT10A | chr2 | 219,744,145 | 219,745,748 | 9 | 2.17 × 10−5 | 0.0113 | 0.83 | 0.0307 | 33.3% |
| CRISP2 | chr6 | 49,681,178 | 49,681,774 | 11 | 2.19 × 10−5 | 0.0113 | 0.89 | 0.0128 | 100.0% |
| RBPJL | chr20 | 43,934,854 | 43,935,551 | 12 | 2.20 × 10−5 | 0.0113 | 0.72 | 0.1048 | 66.7% |
| KDM2B | chr12 | 122,018,574 | 122,020,205 | 14 | 2.21 × 10−5 | 0.0113 | 0.83 | 0.0336 | 50.0% |
| IL27 | chr16 | 28,518,114 | 28,519,597 | 9 | 4.34 × 10−5 | 0.0156 | 0.75 | 0.0844 | 33.3% |
| CA14 | chr1 | 150,229,143 | 150,230,345 | 9 | 6.51 × 10−5 | 0.0196 | 0.78 | 0.0585 | 33.3% |
| C9orf47 | chr9 | 91,604,473 | 91,606,140 | 12 | 2.64 × 10−4 | 0.0318 | 0.79 | 0.0518 | 18.2% |
| STAB1 | chr3 | 52,528,714 | 52,529,393 | 8 | 3.04 × 10−4 | 0.0329 | 0.79 | 0.0534 | 12.5% |
| ACY3 | chr11 | 67,415,183 | 67,418,365 | 8 | 3.26 × 10−4 | 0.0329 | 0.72 | 0.1045 | 62.5% |
| MPG | chr16 | 125,896 | 128,009 | 11 | 3.29 × 10−4 | 0.0329 | 0.82 | 0.0368 | 10.0% |
| ESM1 | chr5 | 54,281,198 | 54,282,459 | 13 | 3.97 × 10−4 | 0.0360 | 0.79 | 0.0526 | 61.5% |
| TMEM219 | chr16 | 29,972,752 | 29,974,294 | 6 | 5.57 × 10−4 | 0.0431 | 0.77 | 0.0667 | 66.7% |
| CREBBP | chr16 | 3,930,112 | 3,931,489 | 5 | 6.16 × 10−4 | 0.0457 | 0.76 | 0.0725 | 80.0% |
| Severity DMR Analysis | |||||||||
| GANC | chr15 | 42,565,522 | 42,566,390 | 7 | 3.38 × 10−4 | 0.0357 | 0.88 | 0.0153 | 28.6% |
| NME3 | chr16 | 1,821,559 | 1,822,346 | 8 | 3.59 × 10−4 | 0.0366 | 0.76 | 0.0729 | 37.5% |
| SLC6A5 | chr11 | 20,619,598 | 20,621,109 | 8 | 3.59 × 10−4 | 0.0366 | 0.80 | 0.0489 | 28.6% |
| RAB22A | chr20 | 56,883,532 | 56,885,003 | 8 | 4.38 × 10−4 | 0.0391 | 0.78 | 0.0594 | 25.0% |
| ACTN4 | chr19 | 39,137,911 | 39,138,334 | 7 | 4.89 × 10−4 | 0.0407 | 0.84 | 0.0298 | 33.3% |
| NPY | chr7 | 24,322,873 | 24,324,570 | 8 | 5.58 × 10−4 | 0.0421 | 0.84 | 0.0276 | 37.5% |
| RAB38 | chr11 | 87,908,558 | 87,909,729 | 9 | 6.33 × 10−4 | 0.045 | 0.73 | 0.0995 | 44.4% |
| COPB1 | chr11 | 14,521,639 | 14,522,617 | 6 | 7.79 × 10−4 | 0.0495 | 0.88 | 0.0149 | 50.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carry, P.M.; Terhune, E.A.; Trahan, G.D.; Vanderlinden, L.A.; Wethey, C.I.; Ebrahimi, P.; McGuigan, F.; Åkesson, K.; Hadley-Miller, N. Severity of Idiopathic Scoliosis Is Associated with Differential Methylation: An Epigenome-Wide Association Study of Monozygotic Twins with Idiopathic Scoliosis. Genes 2021, 12, 1191. https://doi.org/10.3390/genes12081191
Carry PM, Terhune EA, Trahan GD, Vanderlinden LA, Wethey CI, Ebrahimi P, McGuigan F, Åkesson K, Hadley-Miller N. Severity of Idiopathic Scoliosis Is Associated with Differential Methylation: An Epigenome-Wide Association Study of Monozygotic Twins with Idiopathic Scoliosis. Genes. 2021; 12(8):1191. https://doi.org/10.3390/genes12081191
Chicago/Turabian StyleCarry, Patrick M., Elizabeth A. Terhune, George D. Trahan, Lauren A. Vanderlinden, Cambria I. Wethey, Parvaneh Ebrahimi, Fiona McGuigan, Kristina Åkesson, and Nancy Hadley-Miller. 2021. "Severity of Idiopathic Scoliosis Is Associated with Differential Methylation: An Epigenome-Wide Association Study of Monozygotic Twins with Idiopathic Scoliosis" Genes 12, no. 8: 1191. https://doi.org/10.3390/genes12081191
APA StyleCarry, P. M., Terhune, E. A., Trahan, G. D., Vanderlinden, L. A., Wethey, C. I., Ebrahimi, P., McGuigan, F., Åkesson, K., & Hadley-Miller, N. (2021). Severity of Idiopathic Scoliosis Is Associated with Differential Methylation: An Epigenome-Wide Association Study of Monozygotic Twins with Idiopathic Scoliosis. Genes, 12(8), 1191. https://doi.org/10.3390/genes12081191