
Novel FGFR1 Variants Are Associated with Congenital Scoliosis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Bioinformatic Analysis and Mutation Interpretation
- (1)
- Truncating (nonsense, frameshift, splice acceptor/donor) variants or missense variants/inframe indels with a CADD score ≥ 20;
- (2)
- Absent from population genomic databases listed above.
2.3. Site-Directed Mutagenesis
2.4. Receptor Expression and Maturation Studies
2.4.1. Endoglycosidase Digestion
2.4.2. Western Analysis
2.4.3. FGF Reporter Gene Assay
2.4.4. Statistical Analyses
3. Results
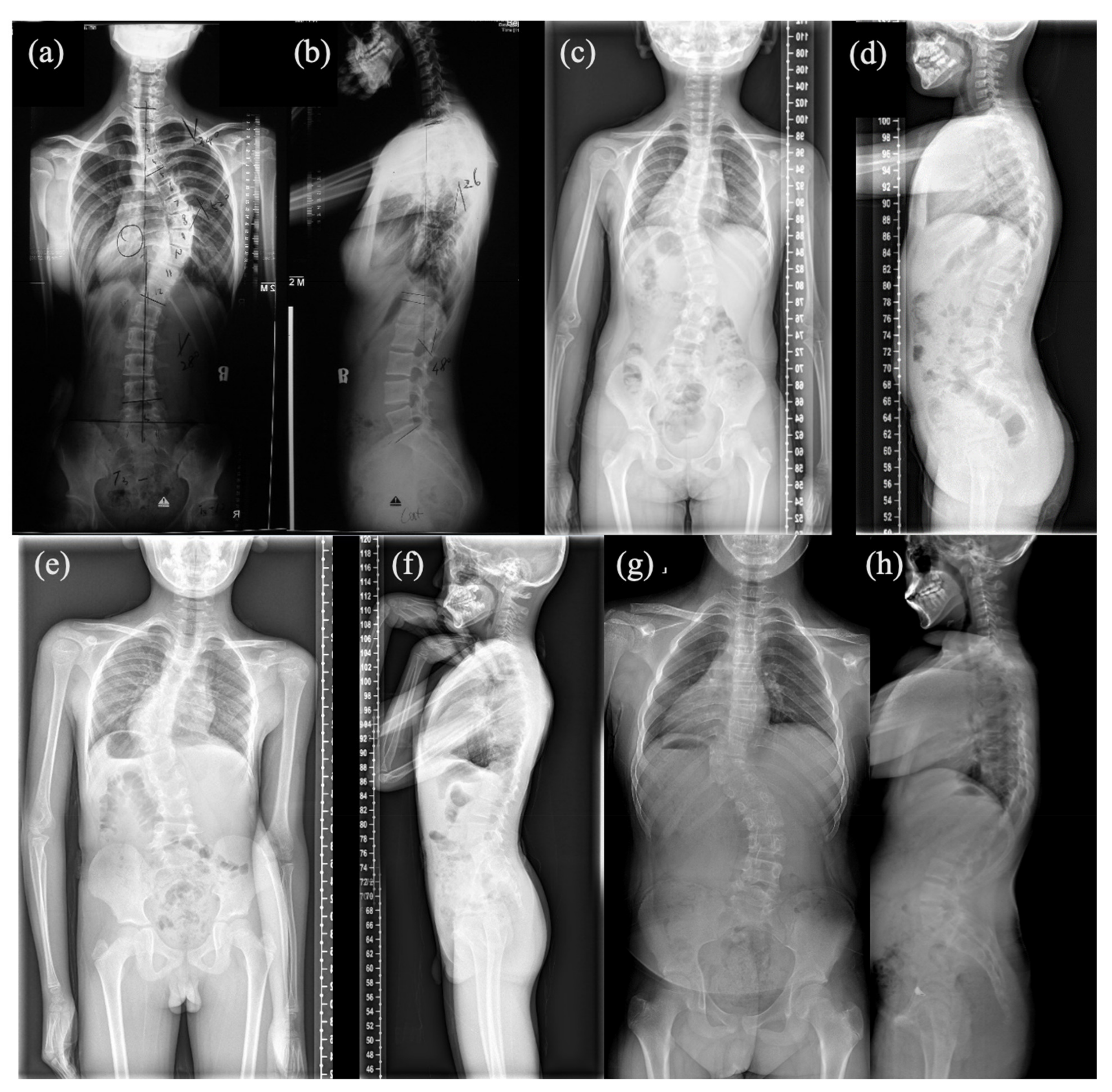
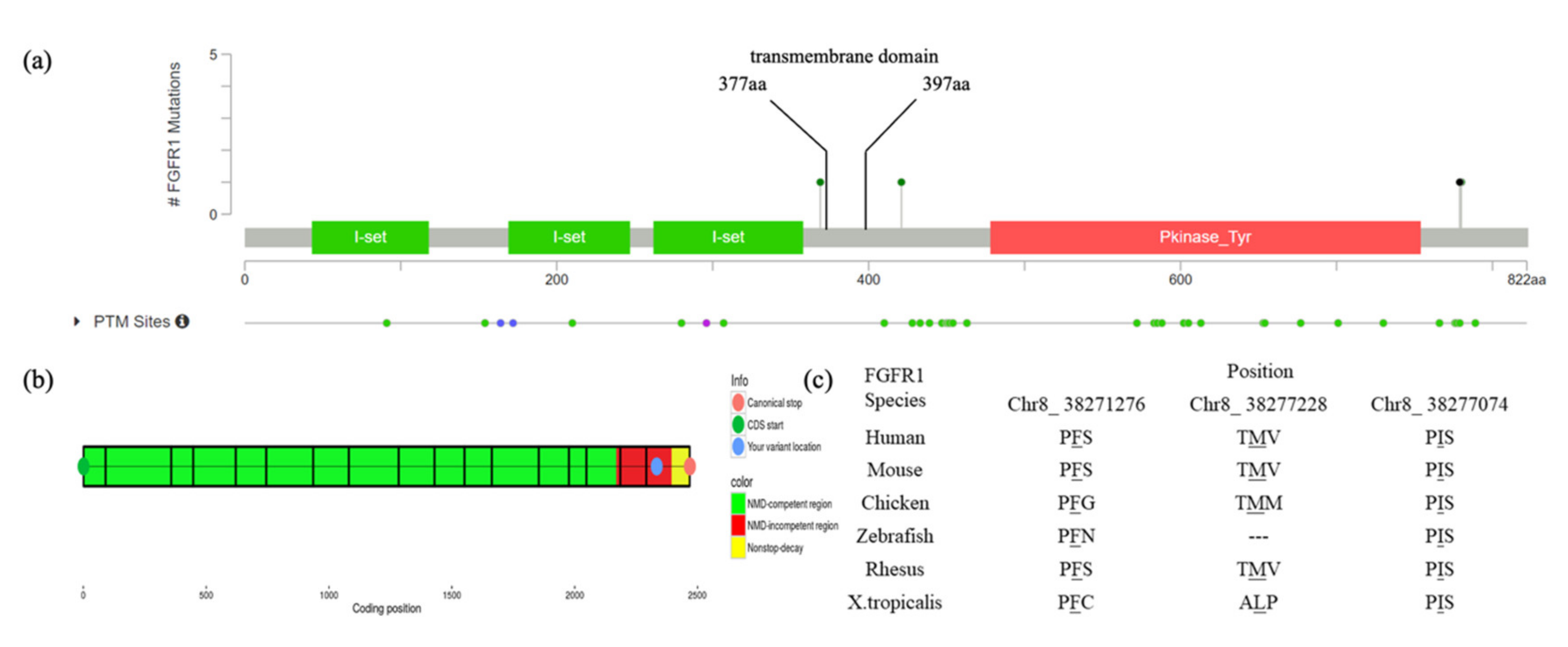
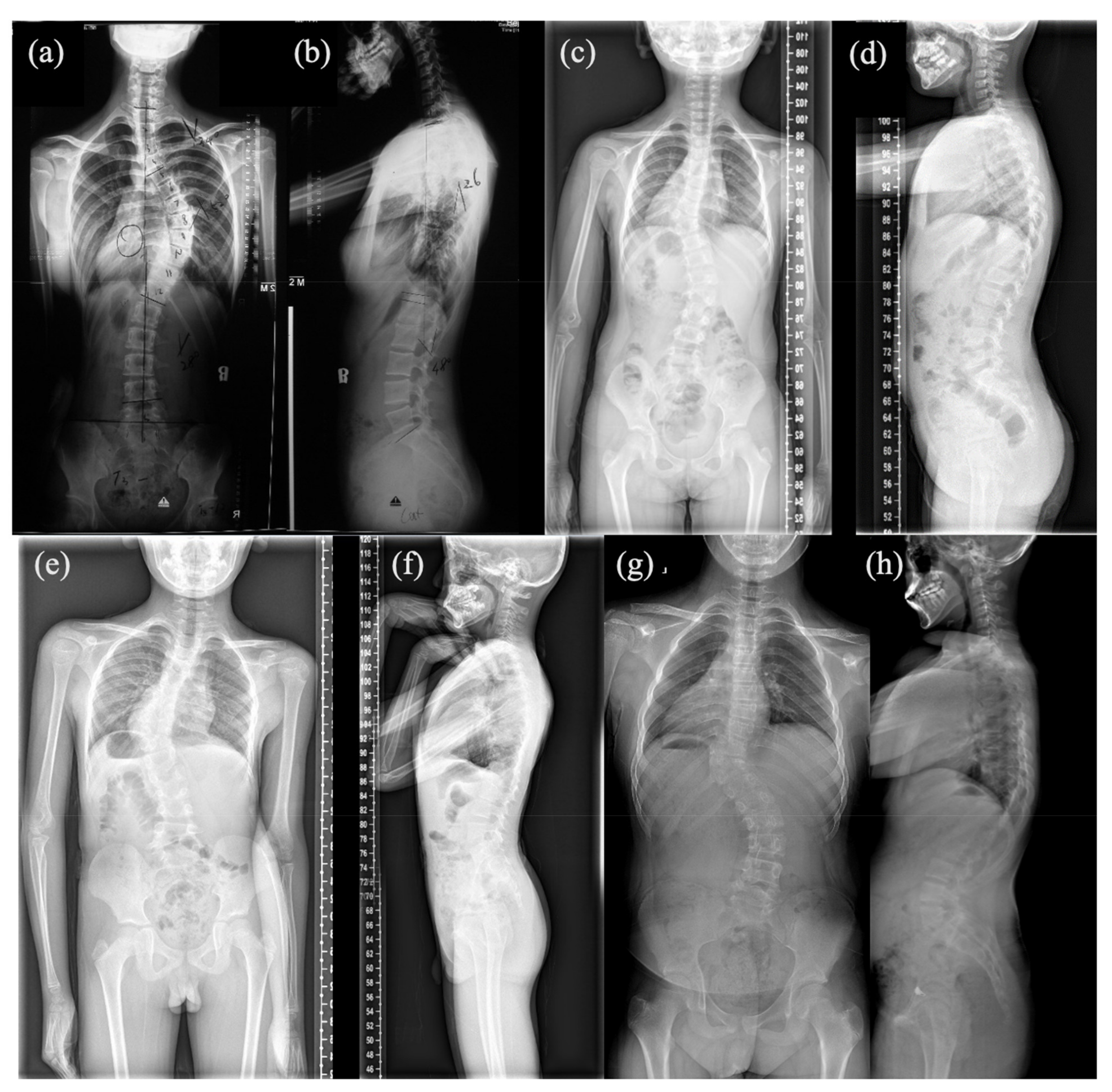
3.1. Mutation and Phenotype Analyses
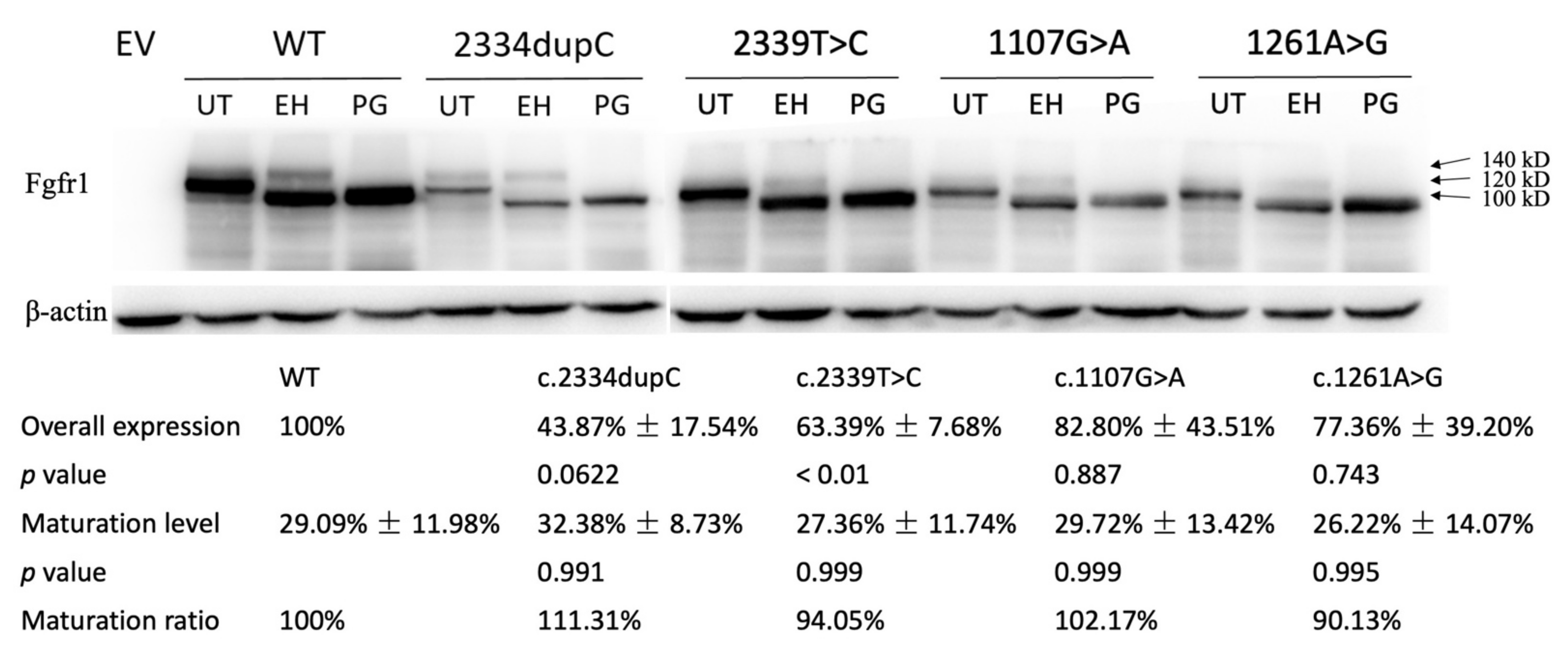
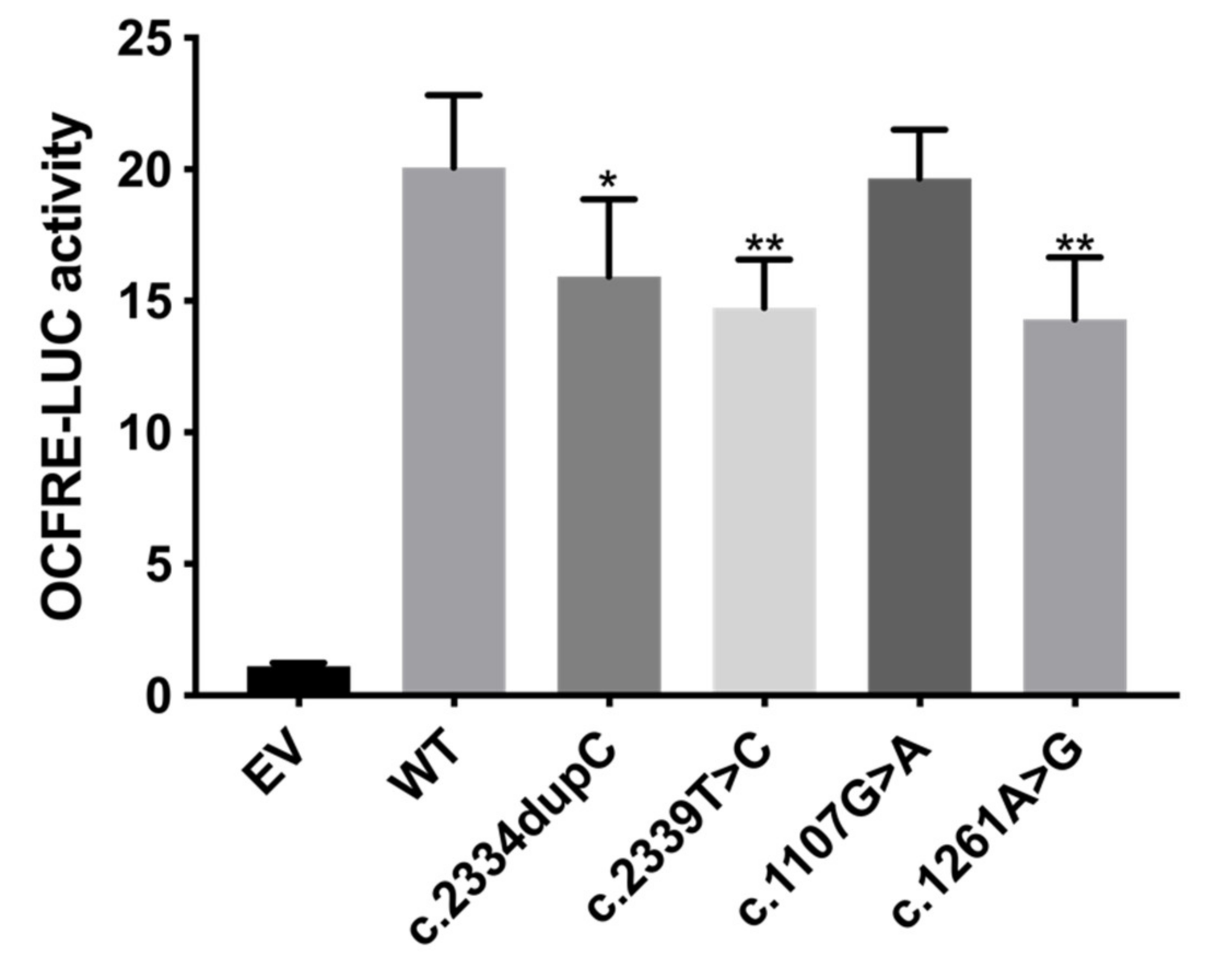
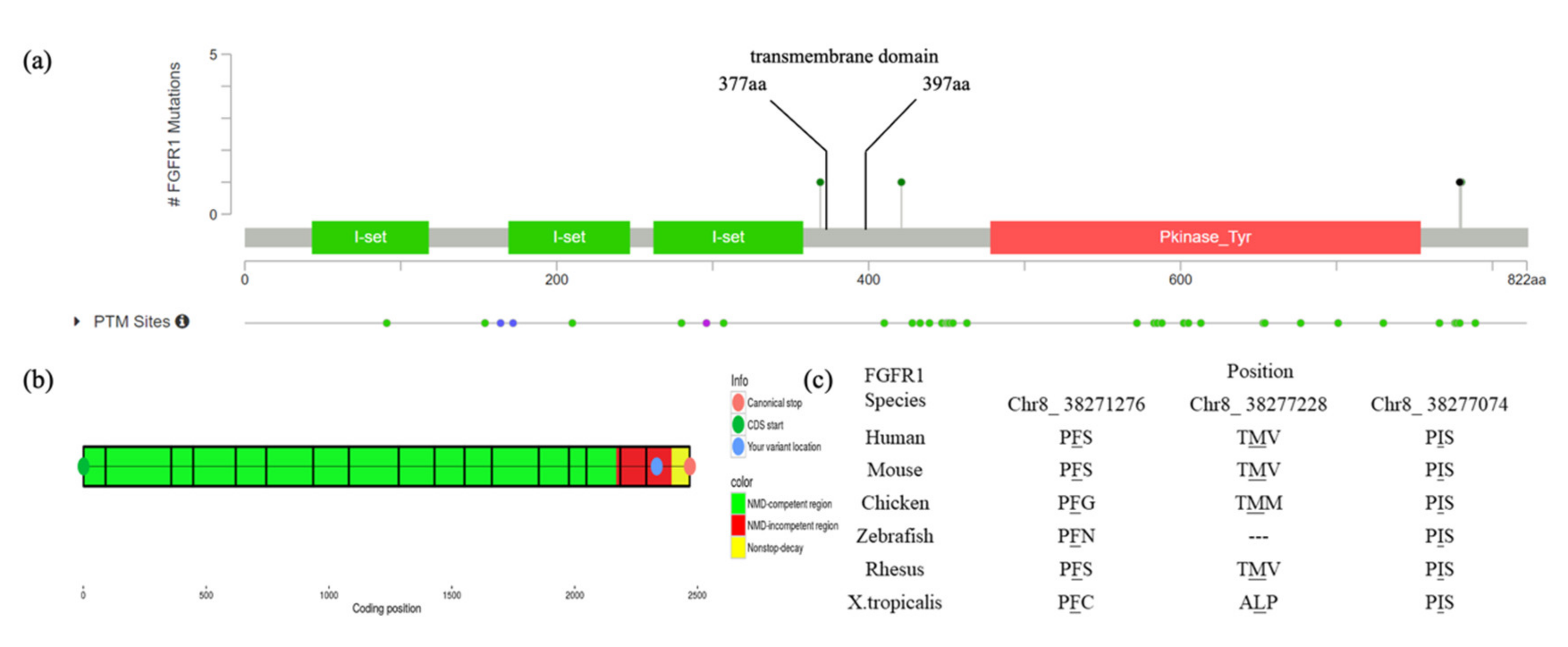
3.2. Functional Characterization of FGFR1 Variants
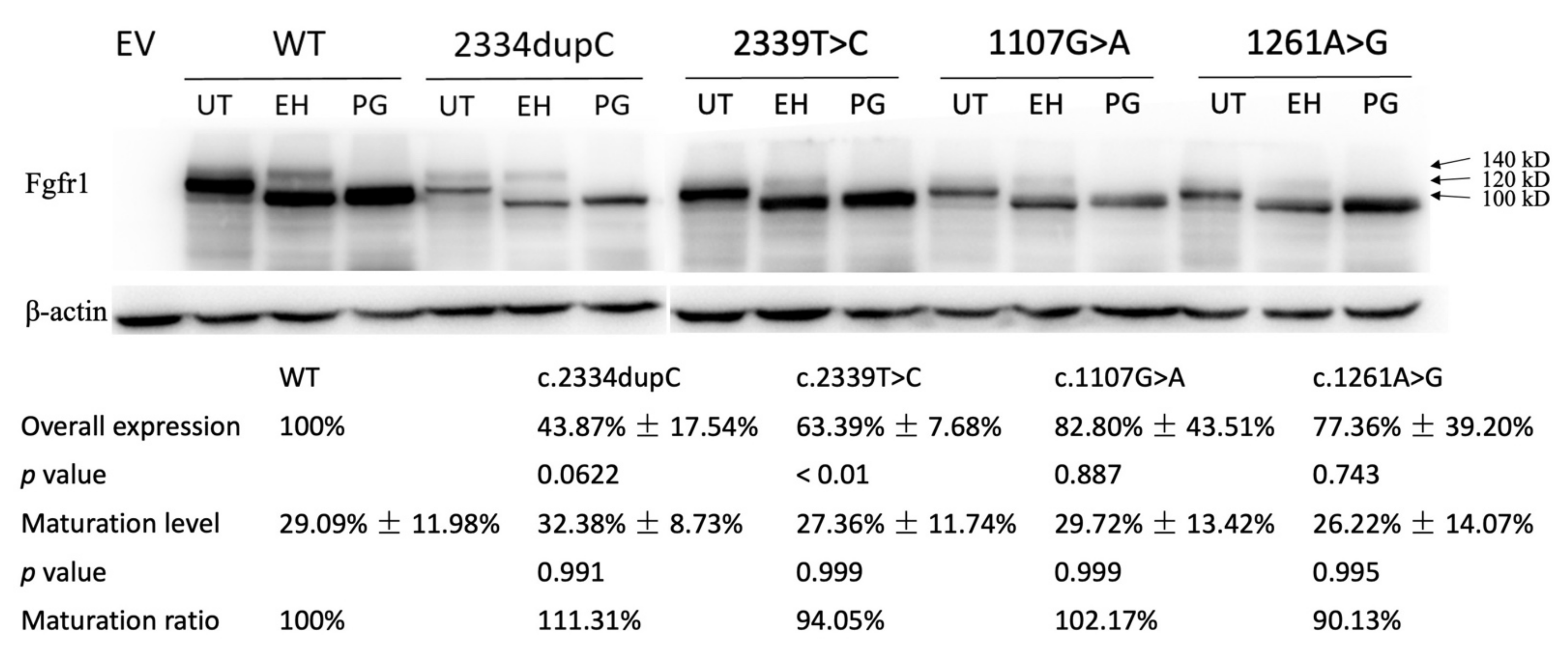
3.2.1. Western Analysis
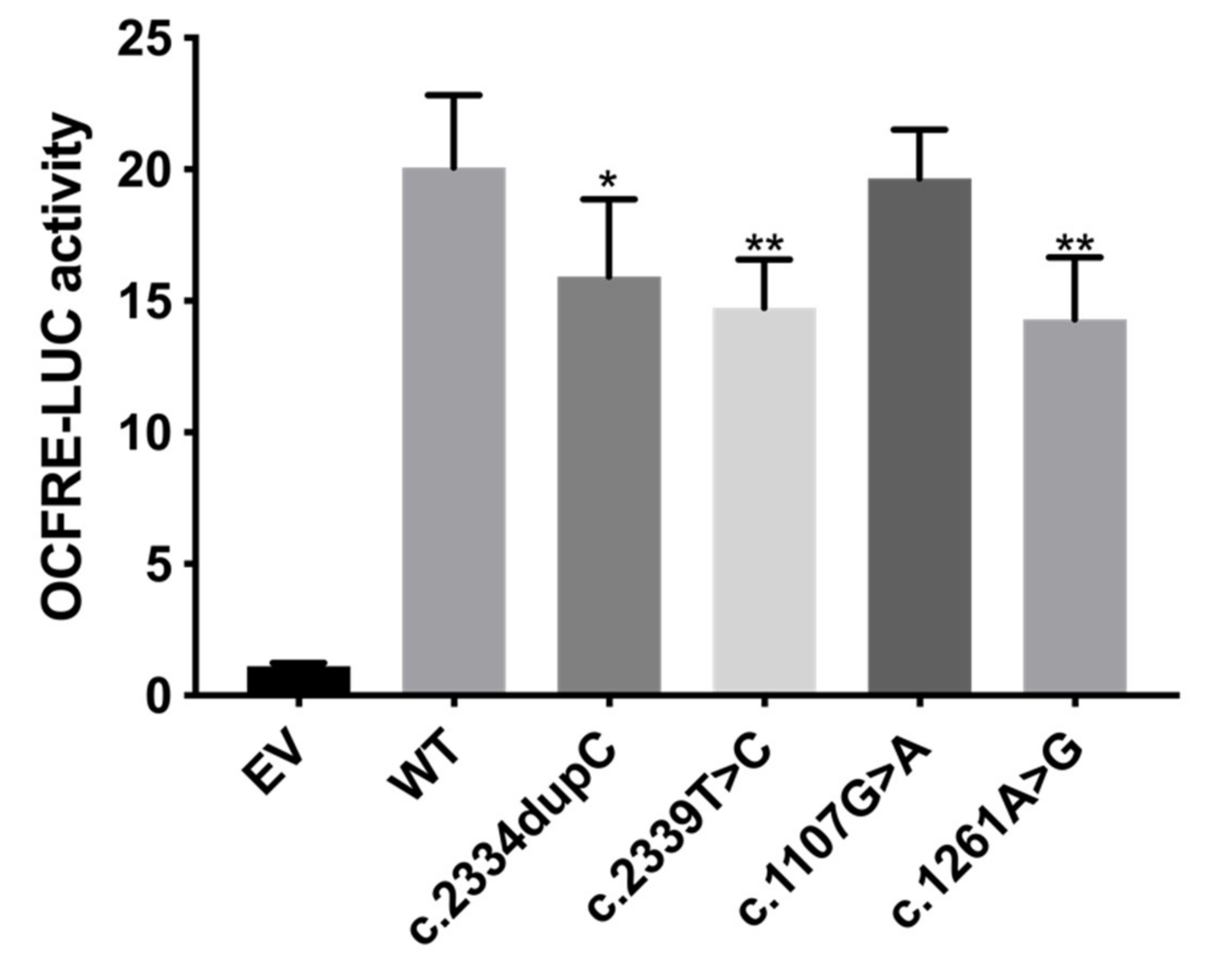
3.2.2. FGF Reporter Gene Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137. [Google Scholar] [CrossRef]
- Partanen, J.; Schwartz, L.; Rossant, J. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev. 1998, 12, 2332–2344. [Google Scholar] [CrossRef] [Green Version]
- Miraoui, H.; Marie, P.J. Fibroblast growth factor receptor signaling crosstalk in skeletogenesis. Sci. Signal. 2010, 3, re9. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, C.; Bastos, M.; Pignatelli, D.; Borges, T.; Aragues, J.M.; Fonseca, F.; Pereira, B.D.; Socorro, S.; Lemos, M.C. Novel FGFR1 mutations in Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism: Evidence for the involvement of an alternatively spliced isoform. Fertil. Steril. 2015, 104, 1261–1267 e1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koika, V.; Varnavas, P.; Valavani, H.; Sidis, Y.; Plummer, L.; Dwyer, A.; Quinton, R.; Kanaka-Gantenbein, C.; Pitteloud, N.; Sertedaki, A.; et al. Comparative functional analysis of two fibroblast growth factor receptor 1 (FGFR1) mutations affecting the same residue (R254W and R254Q) in isolated hypogonadotropic hypogonadism (IHH). Gene 2013, 516, 146–151. [Google Scholar] [CrossRef]
- Ohtaka, K.; Fujisawa, Y.; Takada, F.; Hasegawa, Y.; Miyoshi, T.; Hasegawa, T.; Miyoshi, H.; Kameda, H.; Kurokawa-Seo, M.; Fukami, M.; et al. FGFR1 Analyses in Four Patients with Hypogonadotropic Hypogonadism with Split-Hand/Foot Malformation: Implications for the Promoter Region. Hum. Mutat. 2017, 38, 503–506. [Google Scholar] [CrossRef]
- Raivio, T.; Sidis, Y.; Plummer, L.; Chen, H.; Ma, J.; Mukherjee, A.; Jacobson-Dickman, E.; Quinton, R.; Van Vliet, G.; Lavoie, H.; et al. Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2009, 94, 4380–4390. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Jacobson-Dickman, E.; Xu, C.; Manouvrier, S.; Dwyer, A.A.; Sykiotis, G.P.; Beenken, A.; Liu, Y.; Tommiska, J.; Hu, Y.; et al. Congenital hypogonadotropic hypogonadism with split hand/foot malformation: A clinical entity with a high frequency of FGFR1 mutations. Genet. Med. 2015, 17, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Jarzabek, K.; Wolczynski, S.; Lesniewicz, R.; Plessis, G.; Kottler, M.L. Evidence that FGFR1 loss-of-function mutations may cause variable skeletal malformations in patients with Kallmann syndrome. Adv. Med. Sci. 2012, 57, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.L.; Smith, C.; Partanen, J.; Ornitz, D.M. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev. Biol. 2006, 296, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E.; The Mouse Genome Database Group. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Zhang, Y.; Chen, W.; Li, W.; Wang, S.; Wang, L.; Zhao, Y.; Lin, M.; Ye, Y.; Lin, J.; et al. Diagnostic yield and clinical impact of exome sequencing in early-onset scoliosis (EOS). J. Med. Genet. 2021, 58, 41–47. [Google Scholar] [CrossRef]
- Chen, N.; Zhao, S.; Jolly, A.; Wang, L.; Pan, H.; Yuan, J.; Chen, S.; Koch, A.; Ma, C.; Tian, W.; et al. Perturbations of genes essential for Mullerian duct and Wolffian duct development in Mayer-Rokitansky-Kuster-Hauser syndrome. Am. J. Hum. Genet. 2021, 108, 337–345. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Horton, W.A.; Degnin, C.R. FGFs in endochondral skeletal development. Trends Endocrinol. Metab. TEM 2009, 20, 341–348. [Google Scholar] [CrossRef]
- Hung, I.H.; Schoenwolf, G.C.; Lewandoski, M.; Ornitz, D.M. A combined series of Fgf9 and Fgf18 mutant alleles identifies unique and redundant roles in skeletal development. Dev. Biol. 2016, 411, 72–84. [Google Scholar] [CrossRef]
- Lewandoski, M.; Sun, X.; Martin, G.R. Fgf8 signalling from the AER is essential for normal limb development. Nat. Genet. 2000, 26, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lavine, K.J.; Hung, I.H.; Ornitz, D.M. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev. Biol. 2007, 302, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xu, J.; Colvin, J.S.; Ornitz, D.M. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002, 16, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Ohbayashi, N.; Shibayama, M.; Kurotaki, Y.; Imanishi, M.; Fujimori, T.; Itoh, N.; Takada, S. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002, 16, 870–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Sun, Y.; Wu, H.; Jia, W.; Guan, Q.; He, Z.; Gao, L.; Zhao, J.; Ji, Y.; Li, G.; et al. Posttranslational Modification Defects in Fibroblast Growth Factor Receptor 1 as a Reason for Normosmic Isolated Hypogonadotropic Hypogonadism. Oxid. Med. Cell Longev. 2020, 2020, 2358719. [Google Scholar] [CrossRef] [PubMed]
- Dode, C.; Levilliers, J.; Dupont, J.M.; De Paepe, A.; Le Du, N.; Soussi-Yanicostas, N.; Coimbra, R.S.; Delmaghani, S.; Compain-Nouaille, S.; Baverel, F.; et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003, 33, 463–465. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.P.; Harpal, K.; Henkemeyer, M.; Rossant, J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 1994, 8, 3032–3044. [Google Scholar] [CrossRef] [Green Version]
- Hajihosseini, M.K.; Lalioti, M.D.; Arthaud, S.; Burgar, H.R.; Brown, J.M.; Twigg, S.R.; Wilkie, A.O.; Heath, J.K. Skeletal development is regulated by fibroblast growth factor receptor 1 signalling dynamics. Development 2004, 131, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Brewer, J.R.; Molotkov, A.; Mazot, P.; Hoch, R.V.; Soriano, P. Fgfr1 regulates development through the combinatorial use of signaling proteins. Genes Dev. 2015, 29, 1863–1874. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient #1 | Patient #2 | Patient #3 | Patient #4 | |
|---|---|---|---|---|
| Sex | Female | Female | Male | Male |
| Age of onset | 11 | 4 | 0 | 1 |
| CS type | Failure of segmentation | Mixed defects | Failure of formation | Failure of formation |
| Vertebral malformation | T6-T10 Spine fusion | T9 Hemivertebrae, T8 Butterfly vertebrae | T10 Hemivertebrae | T10 Hemivertebrae |
| Associated anomalies | Mitral valve prolapse; Fusion of 9th and 10th ribs | 9th, 10th and 12th ribs absent | None | None |
| Variant nomenclature | c.2334dupC(p.Ser779GlnfsTer21) | c.2339T>C(p.Phe780Ser) | c.1107G>A(p.Met369Ile) | c.1261A>G(p.Ile421Val) |
| Mutation type | Frameshift | Missense | Missense | Missense |
| Position | Chr8_38271280 | Chr8_38271276 | Chr8_38277228 | Chr8_38277074 |
| 1000G_ASN_AF | 0 | 0 | 0 | 0 |
| gnomAD_EAS_AF | 0 | 0 | 0 | 0 |
| ESP6500_AF | 0 | 0 | 0 | 0 |
| MutationTaster | NA | 1 | 0.999 | 1 |
| SIFT | NA | 0.53 | 0.25 | 0.02 |
| Polyphen2 | NA | 0.948 | 0.174 | 0.481 |
| LRT | NA | 0 | 0 | 0 |
| CADD PHRED-score | 32 | 24.2 | 22.8 | 22.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Chai, X.; Yan, Z.; Zhao, S.; Yang, Y.; Li, X.; Niu, Y.; Lin, G.; Su, Z.; Wu, Z.; et al. Novel FGFR1 Variants Are Associated with Congenital Scoliosis. Genes 2021, 12, 1126. https://doi.org/10.3390/genes12081126
Wang S, Chai X, Yan Z, Zhao S, Yang Y, Li X, Niu Y, Lin G, Su Z, Wu Z, et al. Novel FGFR1 Variants Are Associated with Congenital Scoliosis. Genes. 2021; 12(8):1126. https://doi.org/10.3390/genes12081126
Chicago/Turabian StyleWang, Shengru, Xiran Chai, Zihui Yan, Sen Zhao, Yang Yang, Xiaoxin Li, Yuchen Niu, Guanfeng Lin, Zhe Su, Zhihong Wu, and et al. 2021. "Novel FGFR1 Variants Are Associated with Congenital Scoliosis" Genes 12, no. 8: 1126. https://doi.org/10.3390/genes12081126
APA StyleWang, S., Chai, X., Yan, Z., Zhao, S., Yang, Y., Li, X., Niu, Y., Lin, G., Su, Z., Wu, Z., Zhang, T. J., & Wu, N. (2021). Novel FGFR1 Variants Are Associated with Congenital Scoliosis. Genes, 12(8), 1126. https://doi.org/10.3390/genes12081126