
Transposons-Based Clonal Diversity in Trematode Involves Parts of CR1 (LINE) in Eu- and Heterochromatin

 ,
,
Abstract
1. Introduction
2. Materials and Methods
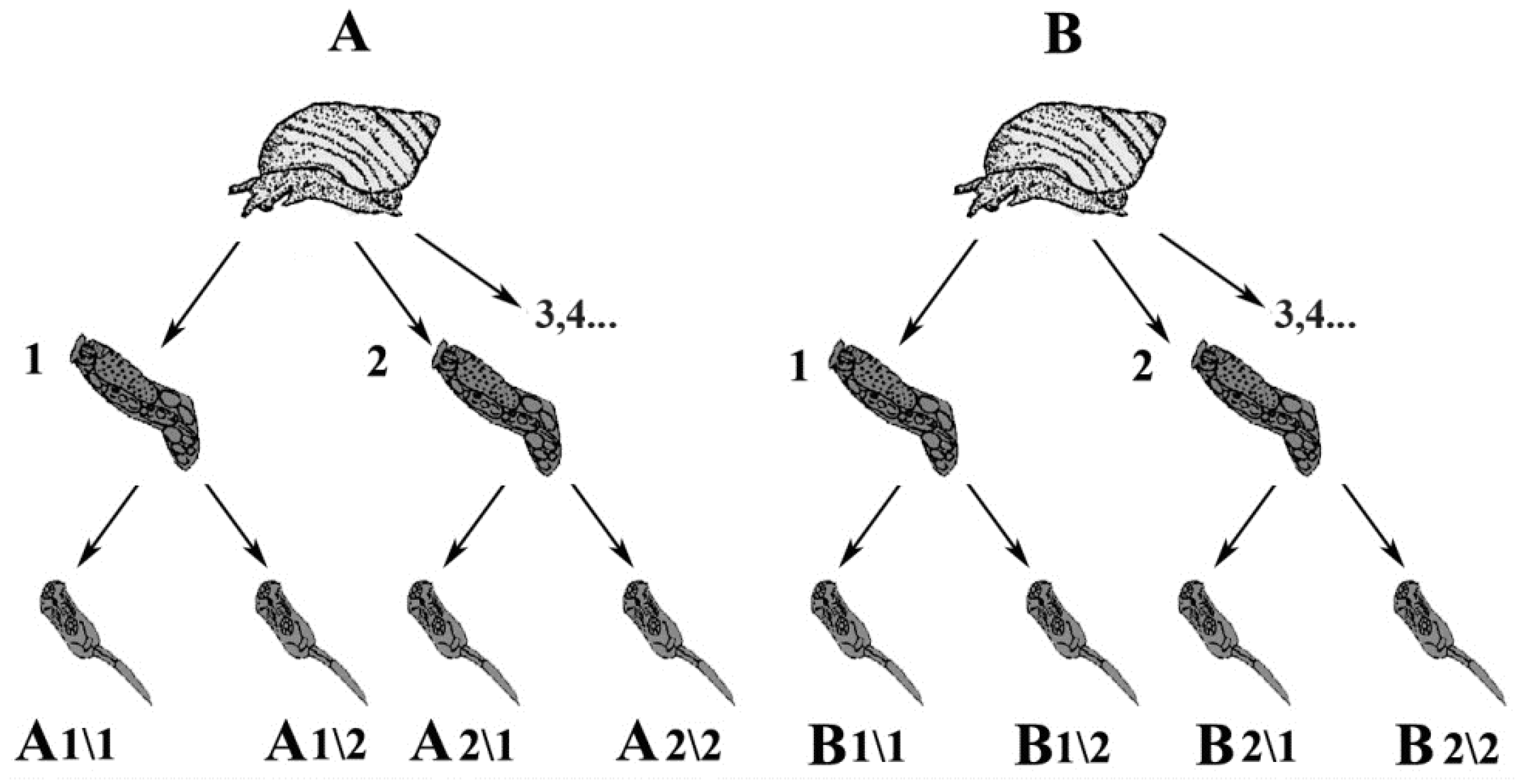
2.1. Trematode Sample Collection
2.2. Primary Genome and Transcriptome Sequencing and Transcriptome Assembly
2.3. Repeat Content Estimation
2.4. SAFLP (Simplified Amplified Fragment Length Polymorphism)
2.5. Fingerprint Visualisation, Cloning and Sequencing
2.6. Cloned Sequence Analysis
2.7. Fluorescence In Situ Hybridisation (FISH)
2.8. Southern Hybridisation
3. Results
3.1. Primary Transcriptome and Partial Genome Sequencing
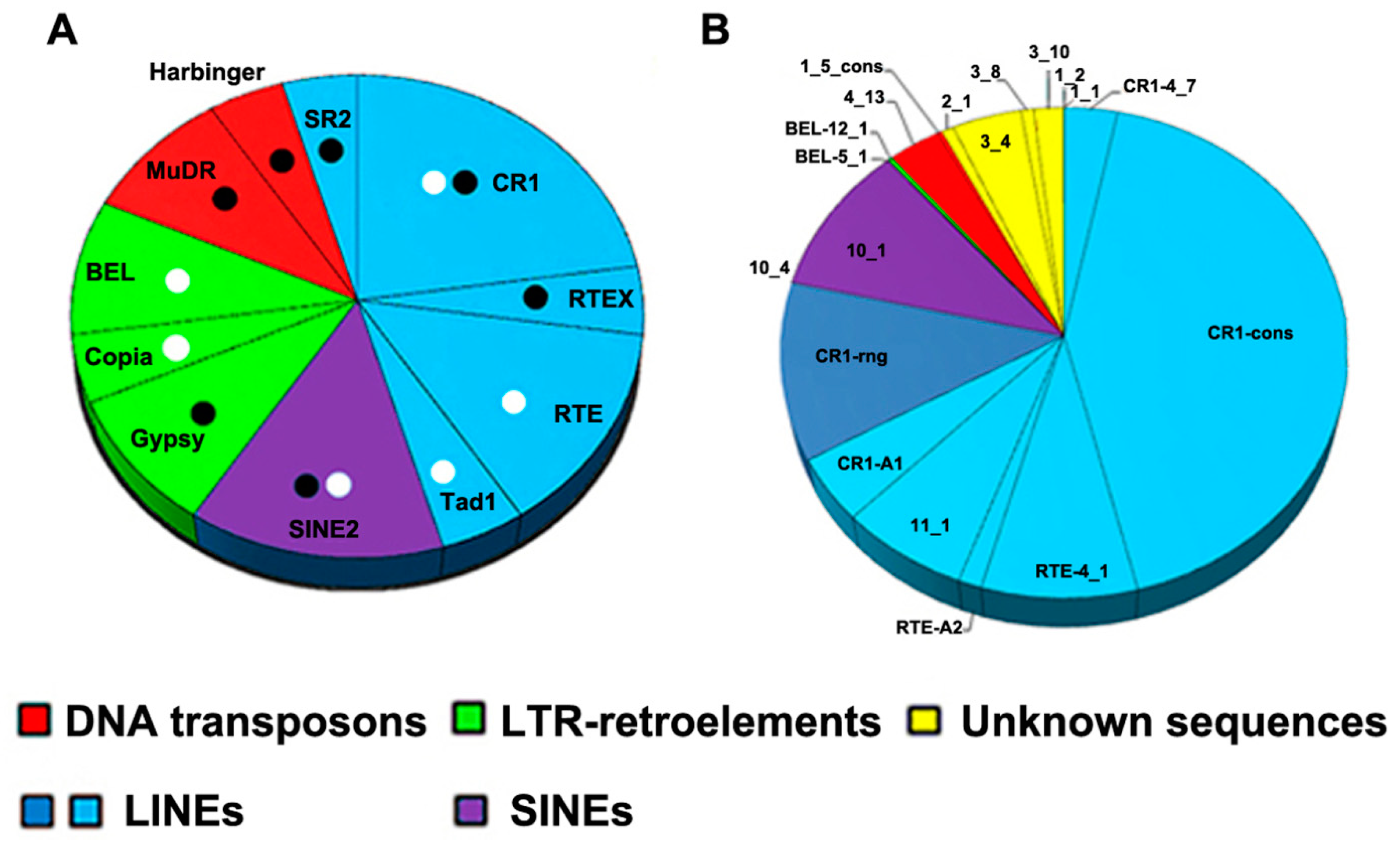
3.2. Repeat Content Analysis
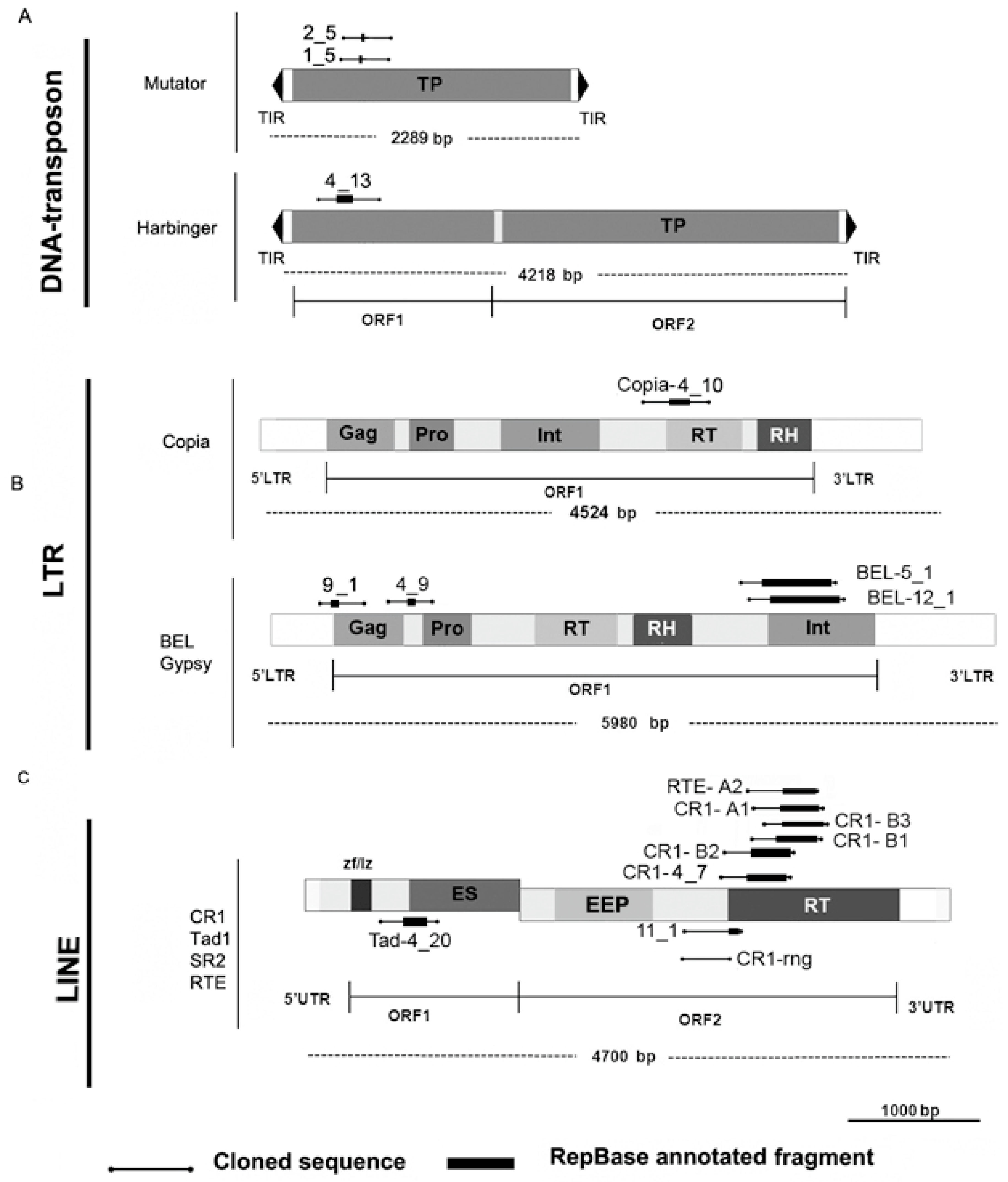
3.3. Description of SAFLP Cloned Fragments
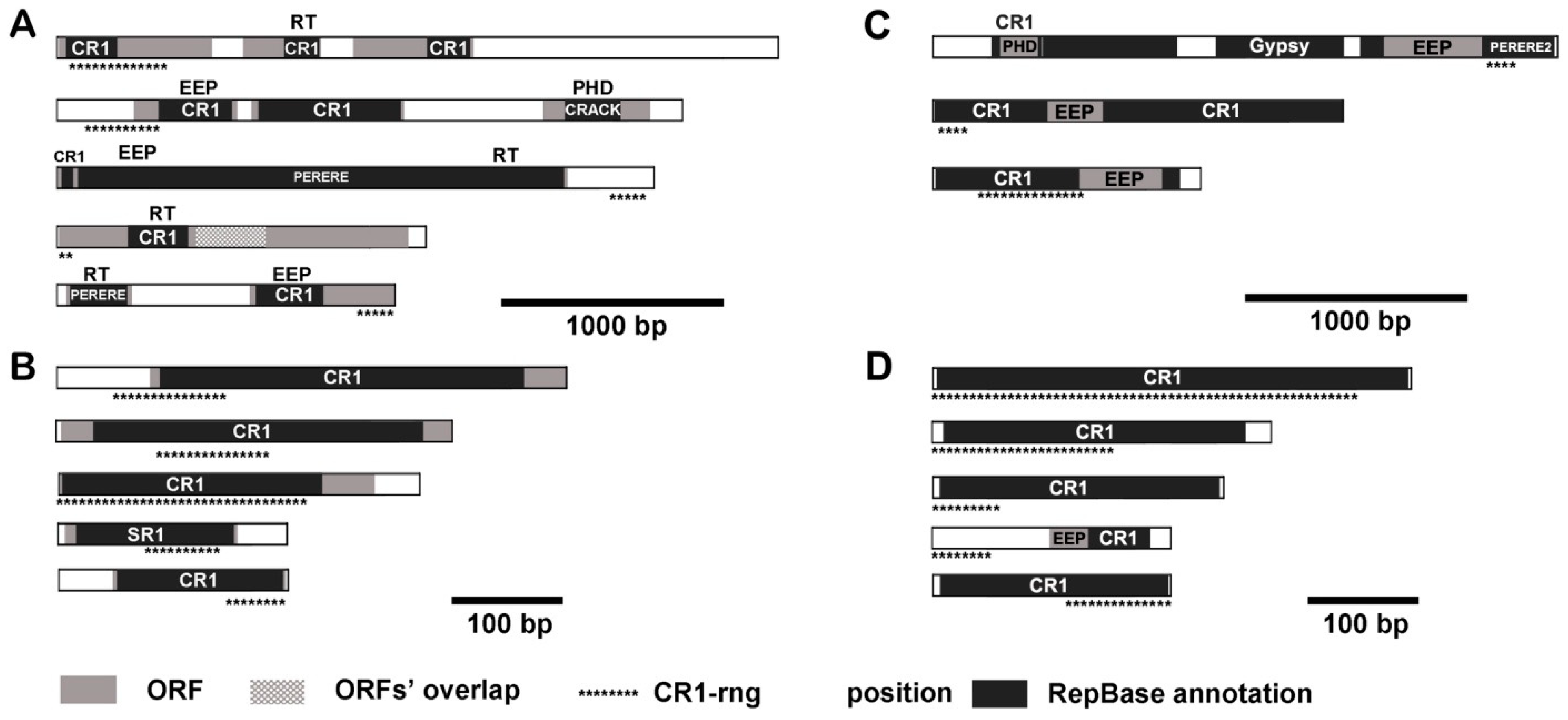
3.4. CR1-rng in Transcripts and Mobilome Contigs
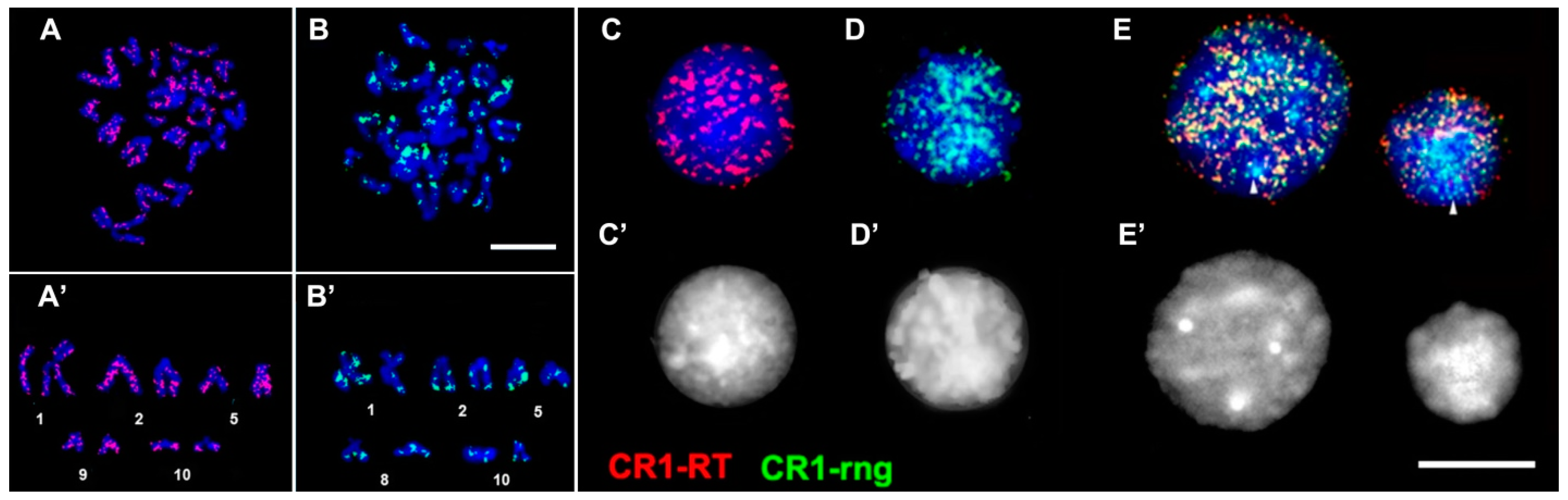
3.5. Physical Mapping of CR1-Like Fragments in the Genome
4. Discussion
4.1. LINEs in Eukaryote Genomes and Transcriptomes
4.2. Trematode Clonal Diversity and LINE Transcription
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.N. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef]
- Edwards, K.J.; Lee, M.; Avramova, Z.; Bennetzen, J.L.; SanMiguel, P.; Tikhonov, A.; Jin, Y.-K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; et al. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar]
- Duret, L.; Marais, G.; Biemont, C. Transposons but not retrotransposons are located preferentially in regions of high recombination rate in Caenorhabditis elegans. Genetics 2000, 156, 1661–1669. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Kaminker, J.S.; Bergman, C.M.; Kronmiller, B.; Carlson, J.; Svirskas, R.; Patel, S.; Frise, E.; Wheeler, D.A.; Lewis, S.E.; Rubin, G.M.; et al. The transposable elements of the Drosophila melanogaster euchromatin: A genomics perspective. Genome Biol. 2002, 3, research0084. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, E.; Buzdin, A. Retroelements and their impact on genome evolution and functioning. Cell. Mol. Life Sci. 2009, 66, 3727–3742. [Google Scholar] [CrossRef]
- Dang-Nguyen, T.Q.; Torres-Padilla, M.E. How cells build totipotency and pluripotency: Nuclear, chromatin and transcriptional architecture. Curr. Opin. Cell Biol. 2015, 34, 9–15. [Google Scholar] [CrossRef]
- Kazazian, H.H. Mobile DNA transposition in somatic cells. BMC Biol. 2011, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Bao, W.; Kojima, K.K. Families of transposable elements, population structure and the origin of species. Biol. Direct 2011, 6, 44. [Google Scholar] [CrossRef]
- Casacuberta, E.; González, J. The impact of transposable elements in environmental adaptation. Mol. Ecol. 2013, 22, 1503–1517. [Google Scholar] [CrossRef]
- Serrato-Capuchina, A.; Matute, D.R. The role of transposable elements in speciation. Genes 2018, 9, 254. [Google Scholar] [CrossRef]
- Dobrovolskij, A.A.; Ataev, G. The nature of reproduction of Trematodes rediae and sporocysts. In Taxonomy, Ecology and Evolution of Metazoan Parasites; Presses Universitaires de Perpignan: Perpignan, France, 2003; Volume 1. [Google Scholar]
- Pearson, J.C. A phylogeny of life-cycle patterns of the Digenea. In Advances in Parasitology; Dawes, B.B.T., Ed.; Academic Press: Cambridge, MA, USA, 1972; Volume 10, pp. 153–189. ISBN 0065-308X. [Google Scholar]
- Webster, P.; Mansour, T.E.; Bieber, D. Isolation of a female-specific, highly repeated Schistosoma mansoni DNA probe and its use in an assay of cercarial sex. Mol. Biochem. Parasitol. 1989, 36, 217–222. [Google Scholar] [CrossRef]
- Drew, A.C.; Brindley, P.J. Female-specific sequences isolated from Schistosoma mansoni by representational difference analysis. Mol. Biochem. Parasitol. 1995, 71, 173–181. [Google Scholar] [CrossRef]
- Grevelding, C.G. Genomic instability in Schistosoma mansoni. Mol. Biochem. Parasitol. 1999, 101, 207–216. [Google Scholar] [CrossRef]
- Bayne, C.J.; Grevelding, C.G. Cloning of Schistosoma mansoni sporocysts in vitro and detection of genetic heterogeneity among individuals within clones. J. Parasitol. 2003, 89, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Khalturin, K.V.; Mikhaĭlova, N.A.; Granovich, A.I. Genetic heterogeneity in natural populations of Microphallus piriformes and M. pygmaeus parthenites (Trematoda: Microphallidae). Parazitologiia 2000, 34, 486–501. [Google Scholar] [PubMed]
- Semyenova, S.K.; Khrisanfova, G.G.; Korsunenko, A.V.; Voronin, M.V.; Beer, S.V.; Vodyanitskaya, S.V.; Serbina, E.A.; Yurlova, N.I.; Ryskov, A.P. Multilocus variation in cercariae, parthenogenetic progeny of different species of the class Trematoda. Dokl. Biol. Sci. 2007, 414, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Hu, W.; Mo, X.; Wang, S.; Brindley, P.J.; McManus, D.P.; Davis, G.M.; Feng, Z.; Blair, D. Multiple near-identical genotypes of Schistosoma japonicum can occur in snails and have implications for population-genetic analyses. Int. J. Parasitol. 2008. [Google Scholar] [CrossRef]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; van de Lee, T.; Hornes, M.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 44, 388–396. [Google Scholar] [CrossRef]
- Mueller, U.G.; Wolfenbarger, L.L.R. AFLP genotyping and fingerprinting. Trends Ecol. Evol. 1999, 14, 389–394. [Google Scholar] [CrossRef]
- Behura, S.K. Individual analysis of transposon polymorphisms by AFLP BT —Mobile genetic elements: Protocols and genomic applications. In Mobile Genetic Elements; Bigot, Y., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 155–167. ISBN 978-1-61779-603-6. [Google Scholar]
- Frascaroli, E.; Schrag, T.A.; Melchinger, A.E. Genetic diversity analysis of elite European maize (Zea mays L.) inbred lines using AFLP, SSR, and SNP markers reveals ascertainment bias for a subset of SNPs. Theor. Appl. Genet. 2013, 126, 133–141. [Google Scholar] [CrossRef]
- Galaktionov, N.K.; Solovyeva, A.I.; Fedorov, A.V.; Podgornaya, O.I. Trematode Himasthla elongata mariner element (Hemar): Structure and applications. J. Exp. Zool. Part B Mol. Dev. Evol. 2014, 322, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Levakin, I.A.; Losev, E.A.; Zavirskii, I.V.; Galaktionov, K.V. Clonal variability in longevity of the cercariae of Himasthla elongata (Trematoda: Echinostomatidae). Parazitologiia 2013, 47, 353–360. [Google Scholar] [PubMed]
- Prokof’ev, V.V.; Levakin, I.A.; Losev, E.A.; Zavirinskii, I.V.; Galaktionov, K.V. Clonal variability in expression of geo- and photoorientation in cercariae of Himasthla elongata (Trematoda: Echinostomatidae). Parazitologiia 2011, 45, 345–357. [Google Scholar]
- Galaktionov, N.K.; Podgornaya, O.I.; Strelkov, P.P.; Galaktionov, K.V. Genomic diversity of cercarial clones of Himasthla elongata (Trematoda, Echinostomatidae) determined with AFLP technique. Parasitol. Res. 2016, 115, 4587–4593. [Google Scholar] [CrossRef]
- Solovyeva, A.I.; Galaktionov, N.K.; Podgornaya, O.I. LINE class retroposon is a component of DNA polymorphic fragments of trematode Himasthla elongata parthenitae. Cell Tissue Biol. 2013, 7, 563–572. [Google Scholar] [CrossRef]
- Winnepenninckx, B.; Backeljau, T.; De Wachter, R. Extraction of high molecular weight DNA from molluscs. Trends Genet. 1993, 9, 407. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Przhibelskiy, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. bioRxiv 2018, 420208. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Suvorov, V.; Prjibelski, A.D. rnaQUAST: A quality assessment tool for de novo transcriptome assemblies. Bioinformatics 2016, 32, 2210–2212. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipmanl, D.J. Basic local alignment search tool. J. Mol. Biol. 1990. [Google Scholar] [CrossRef]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 473–479. [Google Scholar] [CrossRef]
- Vassetzky, N.S.; Kramerov, D.A. SINEBase: A database and tool for SINE analysis. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef]
- Hall, T. BioEdit Version 5.0.6; North Carolina State University: Raleigh, NC, USA, 2001; p. 192. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016. [Google Scholar] [CrossRef]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2009, 26, 493–500. [Google Scholar] [CrossRef]
- Solovyeva, A.I.; Stefanova, V.N.; Podgornaya, O.I.; Demin, S.I. Karyotype features of trematode Himasthla elongata. Mol. Cytogenet. 2016, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Wahl, G.M.; Stern, M.; Stark, G.R. Efficient transfer of large DNA fragments from agarose gels to diazobenzyloxymethyl-paper and rapid hybridization by using dextran sulfate. Proc. Natl. Acad. Sci. USA 1979, 76, 3683–3687. [Google Scholar] [CrossRef]
- Leary, J.J.; Brigati, D.J.; Ward, D.C. Rapid and sensitive colorimetric method for visualizing biotin-labeled DNA probes hybridized to DNA or RNA immobilized on nitrocellulose: Bio-blots. Proc. Natl. Acad. Sci. USA 1983, 80, 4045–4049. [Google Scholar] [CrossRef] [PubMed]
- Fadloun, A.; Le Gras, S.; Jost, B.; Ziegler-Birling, C.; Takahashi, H.; Gorab, E.; Carninci, P.; Torres-Padilla, M.E. Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat. Struct. Mol. Biol. 2013, 20, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.A.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.A.; Hughes, D.A.; Hedges, D.J.; Anders, B.A.; Laborde, M.E.; Shewale, J.; Sinha, S.K.; Batzer, M.A. Quantitative PCR for DNA identification based on genome-specific interspersed repetitive elements. Genomics 2004, 83, 518–527. [Google Scholar] [CrossRef]
- Ray, D.A.; Walker, J.A.; Batzer, M.A. Mobile element-based forensic genomics. Mutat. Res./Fundam. Mol. Mech. Mutagenes. 2007, 616, 24–33. [Google Scholar] [CrossRef][Green Version]
- Laha, T.; Brindley, P.J.; Verity, C.K.; Mcmanus, D.P.; Loukas, A. Pido, a non-long terminal repeat retrotransposon of the chicken repeat 1 family from the genome of the Oriental blood fluke, Schistosoma japonicum. Gene 2002, 284, 149–159. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. The esterase and PHD domains in CR1-like non-LTR retrotransposons. Mol. Biol. Evol. 2003, 20, 38–46. [Google Scholar] [CrossRef]
- Koga, A.; Hori, H.; Ishikawa, Y. Gamera, a family of LINE-like repetitive sequences widely distributed in medaka and related fishes. Heredity 2002, 89, 446–452. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, R.; Kowaltowski, A.T.; Machado, A.A.; Soares, M.B.; Gargioni, C.; Kawano, T.; Rodrigues, V.; Madeira, A.M.B.N.; Wilson, R.A.; Menck, C.F.M.; et al. Saci-1, -2, and -3 and Perere, four novel retrotransposons with high transcriptional activities from the human parasite Schistosoma mansoni. J. Virol. 2004, 78, 2967–2978. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kuznetsova, I.S.; Prusov, A.N.; Enukashvily, N.I.; Podgornaya, O.I. New types of mouse centromeric satellite DNAs. Chromosome Res. 2005. [Google Scholar] [CrossRef]
- Komissarov, A.S.; Gavrilova, E.V.; Demin, S.J.; Ishov, A.M.; Podgornaya, O.I. Tandemly repeated DNA families in the mouse genome. BMC Genom. 2011, 12, 531. [Google Scholar] [CrossRef] [PubMed]
- Staginnus, C.; Desel, C.; Schmidt, T.; Kahl, G. Assembling a puzzle of dispersed retrotransposable sequences in the genome of chickpea (Cicer arietinum L.). Genome 2010, 53, 1090–1102. [Google Scholar] [CrossRef]
- Lakhotia, S.C. Central dogma, selfish DNA and noncoding Rnas: A historical perspective. Proc. Indian Natl. Sci. Acad. 2018, 84, 415–427. [Google Scholar] [CrossRef]
- Solovei, I.; Kreysing, M.; Lanctôt, C.; Kösem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef]
- Buckley, R.M.; Kortschak, R.D.; Raison, J.M.; Adelson, D.L. Similar evolutionary trajectories for retrotransposon accumulation in mammals. Genome Biol. Evol. 2017. [Google Scholar] [CrossRef]
- Ostromyshenskii, D.I.; Chernyaeva, E.N.; Kuznetsova, I.S.; Podgornaya, O.I. Mouse chromocenters DNA content: Sequencing and in silico analysis. BMC Genom. 2018, 19. [Google Scholar] [CrossRef]
- Kuznetsova, I.S.; Ostromyshenskii, D.I.; Komissarov, A.S.; Prusov, A.N.; Waisertreiger, I.S.; Gorbunova, A.V.; Trifonov, V.A.; Ferguson-Smith, M.A.; Podgornaya, O.I. LINE-related component of mouse heterochromatin and complex chromocenters’ composition. Chromosome Res. 2016, 24, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Leung, F.C. A CR1 element is embedded in a novel tandem repeat (HinfI repeat) within the chicken genome. Genome 2006, 49, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Ivancevic, A.M.; Kortschak, R.D.; Bertozzi, T.; Adelson, D.L. LINEs between species: Evolutionary dynamics of LINE-1 retrotransposons across the eukaryotic tree of life. Genome Biol. Evol. 2016, 8, 3301–3322. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Burch, J.B. Evidence that chicken CR1 elements represent a novel family of retroposons. Mol. Cell. Biol. 1989, 9, 3563–3566. [Google Scholar] [CrossRef]
- Haas, N.B.; Grabowski, J.M.; Sivitz, A.B.; Burch, J.B.E. Chicken repeat 1 (CR1) elements, which define an ancient family of vertebrate non-LTR retrotransposons, contain two closely spaced open reading frames. Gene 1997, 197, 305–309. [Google Scholar] [CrossRef]
- Wicker, T.; Robertson, J.S.; Schulze, S.R.; Feltus, F.A.; Magrini, V.; Morrison, J.A.; Mardis, E.R.; Wilson, R.K.; Peterson, D.G.; Paterson, A.H.; et al. The repetitive landscape of the chicken genome. Genome Res. 2005, 15, 126–136. [Google Scholar] [CrossRef]
- Malik, H.S.; Eickbush, T.H. The RTE class of non-LTR retrotransposons is widely distributed in animals and is the origin of many SINEs. Mol. Biol. Evol. 1998, 15, 1123–1134. [Google Scholar] [CrossRef][Green Version]
- Rho, M.; Tang, H. MGEScan-non-LTR: Computational identification and classification of autonomous non-LTR retrotransposons in eukaryotic genomes. Nucleic Acids Res. 2009, 37. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.T.; Mazzuchelli, J.; Ferreira, I.A.; Poletto, A.B.; Fantinatti, B.E.A.; Martins, C. Cytogenetic mapping of the retroelements Rex1, Rex3 and Rex6 among cichlid Fish: New insights on the chromosomal distribution of transposable elements. Cytogenet. Genome Res. 2011, 133, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.M.Z.A.; Pansonato-Alves, J.C.; Utsunomia, R.; Daniel, S.N.; Hashimoto, D.T.; Oliveira, C.; Porto-Foresti, F.; Foresti, F. Chromosomal organization of repetitive DNA sequences in Astyanax bockmanni (Teleostei, Characiformes): Dispersive location, association and co-localization in the genome. Genetica 2013, 141, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Pansonato-Alves, J.C.; Serrano, É.A.; Utsunomia, R.; Scacchetti, P.C.; Oliveira, C.; Foresti, F. Mapping five repetitive DNA classes in sympatric species of Hypostomus (Teleostei: Siluriformes: Loricariidae): Analysis of chromosomal variability. Rev. Fish Biol. Fish. 2013, 23, 477–489. [Google Scholar] [CrossRef]
- Carvalho, N.D.M.; Pinheiro, V.S.S.; Carmo, E.J.; Goll, L.G.; Schneider, C.H.; Gross, M.C. The organization of repetitive DNA in the genomes of amazonian lizard species in the family teiidae. Cytogenet. Genome Res. 2016, 147, 161–168. [Google Scholar] [CrossRef]
- Fischer, C.; Bouneau, L.; Coutanceau, J.P.; Weissenbach, J.; Volff, J.N.; Ozouf-Costaz, C. Global heterochromatic colocalization of transposable elements with minisatellites in the compact genome of the pufferfish Tetraodon nigroviridis. Gene 2004, 336, 175–183. [Google Scholar] [CrossRef]
- Ozouf-Costaz, C.; Brandt, J.; Körting, C.; Pisano, E.; Bonillo, C.; Coutanceau, J.P.; Volff, J.N. Genome dynamics and chromosomal localization of the non-LTR retrotransposons Rex1 and Rex3 in Antarctic fish. Antarct. Sci. 2004, 16, 51–57. [Google Scholar] [CrossRef]
- Suntronpong, A.; Thapana, W.; Twilprawat, P.; Prakhongcheep, O.; Somyong, S.; Muangmai, N.; Peyachoknagul, S.; Srikulnath, K. Karyological characterization and identification of four repetitive element groups (the 18S-28S rRNA gene, telomeric sequences, microsatellite repeat motifs, Rex retroelements) of the Asian swamp eel (Monopterus albus). Comp. Cytogenet. 2017, 11, 435–462. [Google Scholar] [CrossRef] [PubMed]
- Carducci, F.; Barucca, M.; Canapa, A.; Biscotti, M. Rex retroelements and teleost genomes: An overview. Int. J. Mol. Sci. 2018, 19, 3653. [Google Scholar] [CrossRef] [PubMed]
- Valentim, C.L.; Gomes, M.S.; Jeremias, W.J.; Cunha, J.C.; Oliveira, G.C.; Botelho, A.C.; Pimenta, P.F.; Janotti-Passos, L.K.; Guerra-Sa, R.; Baba, E.H. Physical localization of the retrotransposons Boudicca and Perere 03 in Schistosoma mansoni. J. Parasitol. 2008, 94, 993–995. [Google Scholar] [CrossRef]
- Kuris, A.M.; Lafferty, K.D. Community structure: Larval trematodes in snail hosts. Annu. Rev. Ecol. Syst. 1994, 25, 189–217. [Google Scholar] [CrossRef]
- Korsunenko, A.; Chrisanfova, G.; Arifov, A.; Ryskov, A.; Semyenova, S. Characterization of randomly amplified polymorphic DNA (RAPD) fragments revealing clonal variability in cercariae of avian schistosome Trichobilharzia szidati (Trematoda: Schistosomatidae). Open J. Genet. 2013, 2013, 141–158. [Google Scholar] [CrossRef]
- Russo, J.; Harrington, A.W.; Steiniger, M. Antisense transcription of retrotransposons in Drosophila: An origin of endogenous small interfering RNA precursors. Genetics 2016. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, R.; MacHado, A.A.; Bisson-Filho, A.W.; Verjovski-Almeida, S. Identification of 18 new transcribed retrotransposons in Schistosoma mansoni. Biochem. Biophys. Res. Commun. 2005, 333, 230–240. [Google Scholar] [CrossRef]
- Wijayawardena, B.K.; DeWoody, J.A.; Minchella, D.J. The genomic proliferation of transposable elements in colonizing populations: Schistosoma mansoni in the new world. Genetica 2015, 143, 287–298. [Google Scholar] [CrossRef]
- Chinwalla, A.T.; Cook, L.L.; Delehaunty, K.D.; Fewell, G.A.; Fulton, L.A.; Fulton, R.S.; Graves, T.A.; Hillier, L.W.; Mardis, E.R.; McPherson, J.D.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence |
|---|---|
| AdHindF | 5′-GACGATGAGTCCTGAG-3′ |
| AdHindR | 5′-AGCTCTCAGGACTCAT-3′ |
| HindIII + c | 5′-GAGTCCTGAGAGCTTC-3′ |
| Hind + cag | 5′-GAGTCCTGAGAGCTTCAG-3′ |
| Repetitive Element | RepeatExplorer2 Results (Predicted Proportion in Genome, %) | RepeatMasker Search in RepeatExplorer2 Assembled Contigs (% in Mobilome Assembly) | Repeat Masker Search in Transcriptome Contigs (% in Assembly) |
|---|---|---|---|
| LINE | 20.85 | 21.75 | 5.27 |
| Penelope | 0.08 | 0.75 | - |
| LTR | 14.28 | 18.75 | 2.77 |
| DNA transposons | - | 1.95 | 0.63 |
| Unclassified repeats | 4.6 | 14.65 | 5.77 |
| Simple repeats | 4.39 | 1.5 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solovyeva, A.; Levakin, I.; Zorin, E.; Adonin, L.; Khotimchenko, Y.; Podgornaya, O. Transposons-Based Clonal Diversity in Trematode Involves Parts of CR1 (LINE) in Eu- and Heterochromatin. Genes 2021, 12, 1129. https://doi.org/10.3390/genes12081129
Solovyeva A, Levakin I, Zorin E, Adonin L, Khotimchenko Y, Podgornaya O. Transposons-Based Clonal Diversity in Trematode Involves Parts of CR1 (LINE) in Eu- and Heterochromatin. Genes. 2021; 12(8):1129. https://doi.org/10.3390/genes12081129
Chicago/Turabian StyleSolovyeva, Anna, Ivan Levakin, Evgeny Zorin, Leonid Adonin, Yuri Khotimchenko, and Olga Podgornaya. 2021. "Transposons-Based Clonal Diversity in Trematode Involves Parts of CR1 (LINE) in Eu- and Heterochromatin" Genes 12, no. 8: 1129. https://doi.org/10.3390/genes12081129
APA StyleSolovyeva, A., Levakin, I., Zorin, E., Adonin, L., Khotimchenko, Y., & Podgornaya, O. (2021). Transposons-Based Clonal Diversity in Trematode Involves Parts of CR1 (LINE) in Eu- and Heterochromatin. Genes, 12(8), 1129. https://doi.org/10.3390/genes12081129