
Allelic Variants in Established Hypopituitarism Genes Expand Our Knowledge of the Phenotypic Spectrum

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Procedures
2.2. Patients
2.3. DNA Extraction
2.4. Filtering Process and Sequencing Analysis
2.5. Bioinformatics Tools to Check Allelic Variant Impact
3. Results
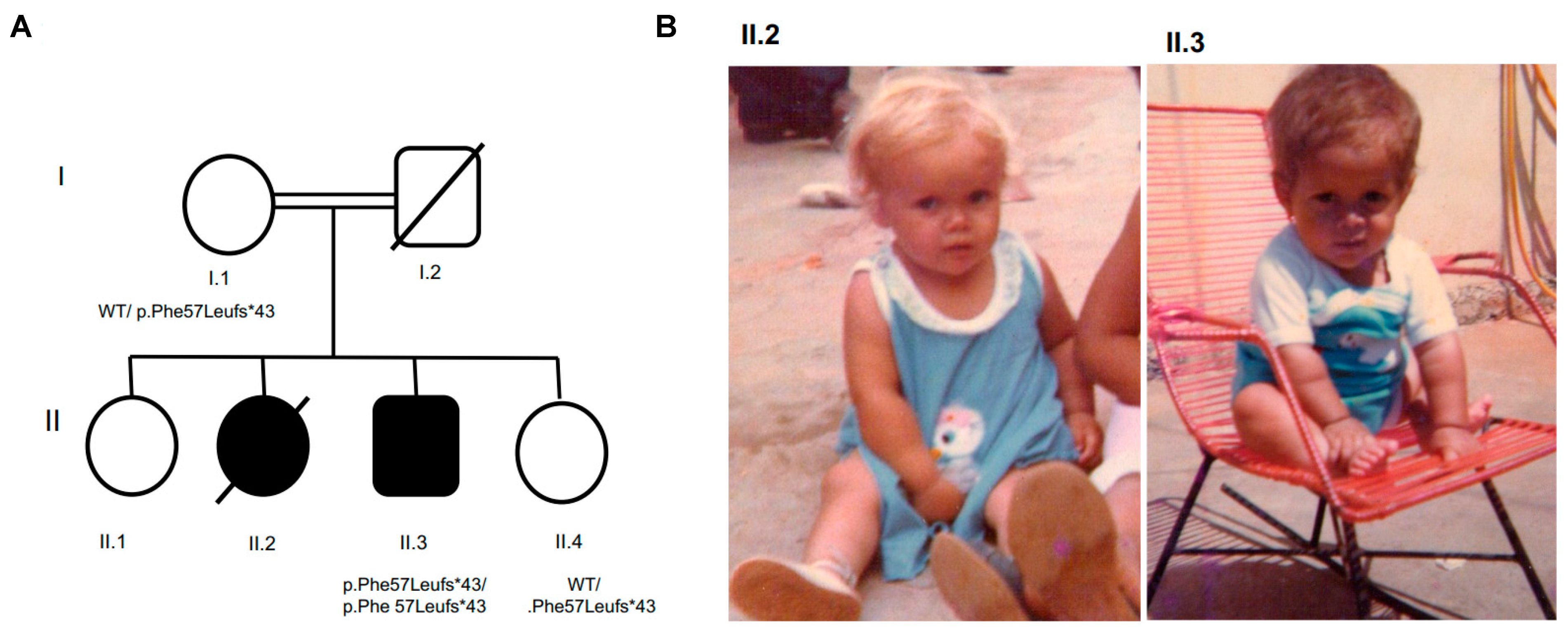
3.1. Patient 1 with Allelic Variant GH1 c.171delT, p.Phe 57Leufs*43, chr17:61995706:delA
3.1.1. Clinical, Laboratory, and Image Features
3.1.2. Molecular Results
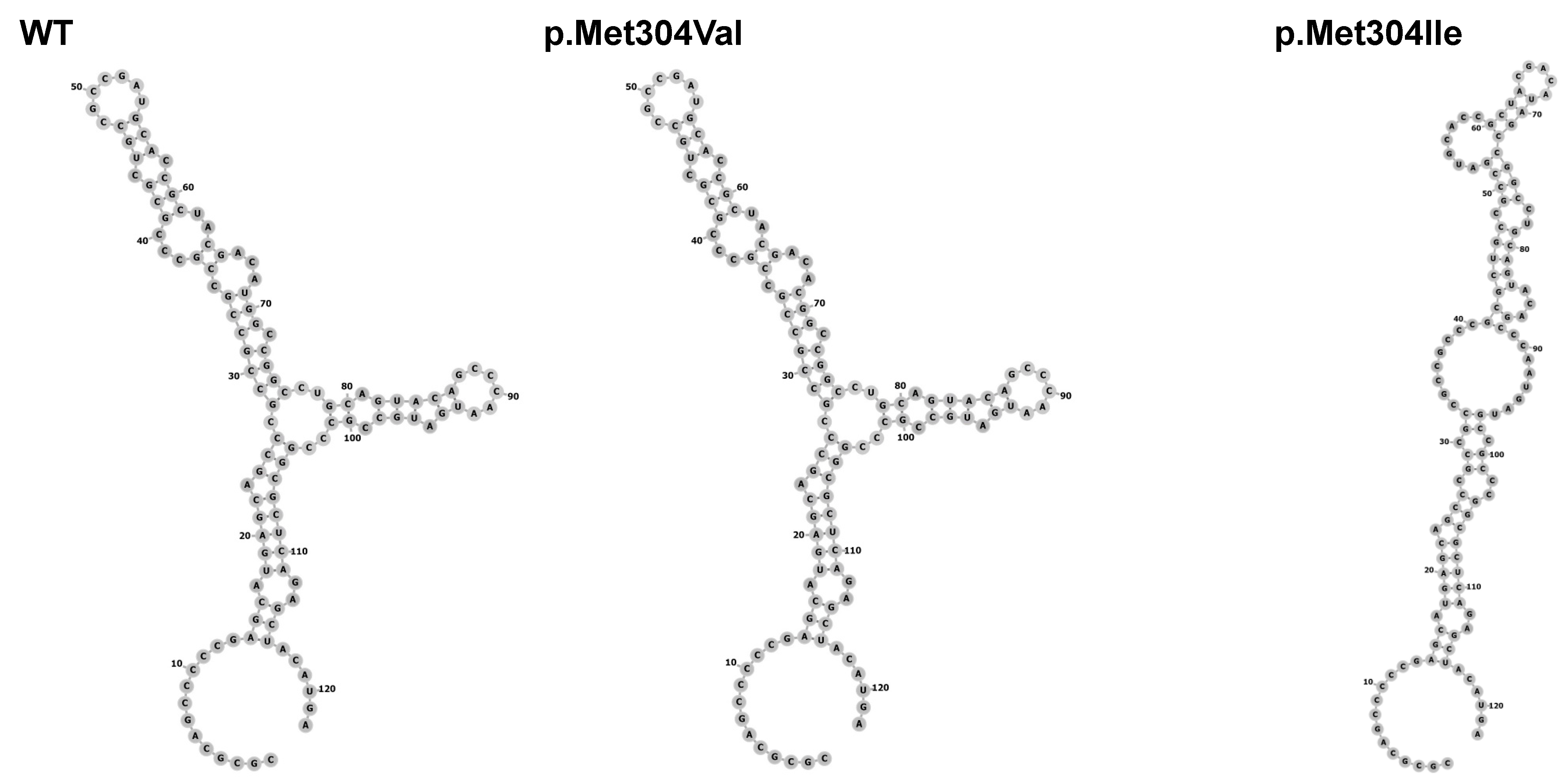
3.2. Patient 2 with Allelic Variant in SOX3 (c.912G>A;p.Met304Ile; chrX:139586314:C>T)
3.2.1. Clinical, Laboratory and Image Features
3.2.2. Molecular Results
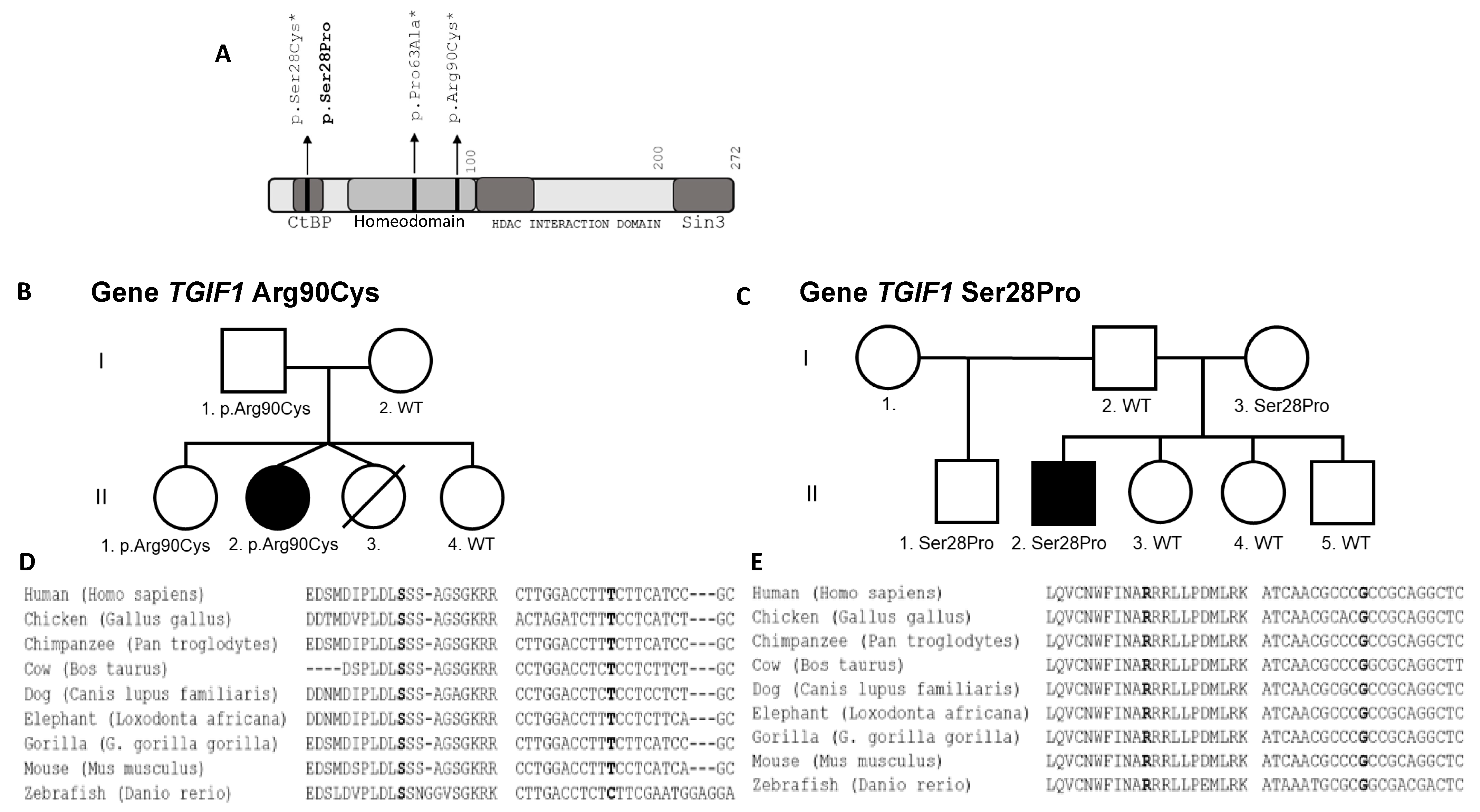
3.3. Patient 3 with Allelic Allelic Variant TGIF1 (c.268C>T;p.Arg90Cys; chr18:3457387:C>T)
3.3.1. Clinical Features and Test Results
3.3.2. Molecular Results
3.4. Patient 4 with Allelic Variant TGIF1 (c.82T>C;p.Ser28Pro; chr18:3456417:T>C)
3.4.1. Clinical Features and Test Results
3.4.2. Molecular Results
4. Discussion
4.1. GH1 Gene
4.2. SOX3 Gene
4.3. TGIF1 Gene
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Castinetti, F.; Reynaud, R.; Saveanu, A.; Quentien, M.H.; Albarel, F.; Barlier, A.; Enjalbert, A.; Brue, T. Clinical and genetic aspects of combined pituitary hormone deficiencies. Ann. Endocrinol. 2008, 69, 7–17. [Google Scholar] [CrossRef]
- Cohen, L.E. Genetic disorders of the pituitary. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; George, A.S.; Brinkmeier, M.L.; Mortensen, A.H.; Gergics, P.; Cheung, L.Y.; Daly, A.Z.; Ajmal, A.; Pérez Millán, M.I.; Ozel, A.B.; et al. Genetics of Combined Pituitary Hormone Deficiency: Roadmap into the Genome Era. Endocr. Rev. 2016, 37, 636–675. [Google Scholar] [CrossRef] [PubMed]
- Otto, A.P.; França, M.M.; Correa, F.A.; Costalonga, E.F.; Leite, C.C.; Mendonca, B.B.; Arnhold, I.J.; Carvalho, L.R.; Jorge, A.A. Frequent development of combined pituitary hormone deficiency in patients initially diagnosed as isolated growth hormone deficiency: A long term follow-up of patients from a single center. Pituitary 2015, 18, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Bashamboo, A.; Bignon-Topalovic, J.; Rouba, H.; McElreavey, K.; Brauner, R. A Nonsense Mutation in the Hedgehog Receptor CDON Associated With Pituitary Stalk Interruption Syndrome. J. Clin. Endocrinol. Metab. 2016, 101, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Giri, D.; Vignola, M.L.; Gualtieri, A.; Scagliotti, V.; McNamara, P.; Peak, M.; Didi, M.; Gaston-Massuet, C.; Senniappan, S. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum. Mol. Genet. 2017, 26, 4315–4326. [Google Scholar] [CrossRef] [PubMed]
- Simm, F.; Griesbeck, A.; Choukair, D.; Weiß, B.; Paramasivam, N.; Klammt, J.; Schlesner, M.; Wiemann, S.; Martinez, C.; Hoffmann, G.F.; et al. Identification of SLC20A1 and SLC15A4 among other genes as potential risk factors for combined pituitary hormone deficiency. Genet. Med. 2018, 20, 728–736. [Google Scholar] [CrossRef]
- Smith, J.D.; Hing, A.V.; Clarke, C.M.; Johnson, N.M.; Perez, F.A.; Park, S.S.; Horst, J.A.; Mecham, B.; Maves, L.; Nickerson, D.A.; et al. Exome sequencing identifies a recurrent de novo ZSWIM6 mutation associated with acromelic frontonasal dysostosis. Am. J. Hum. Genet. 2014, 95, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Gonzalez, M.A.; Lourenco, C.M.; Coutelier, M.; Haack, T.B.; Rebelo, A.; Hannequin, D.; Strom, T.M.; Prokisch, H.; Kernstock, C.; et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014, 137 Pt 1, 69–77. [Google Scholar] [CrossRef]
- Takagi, M.; Narumi, S.; Hamada, R.; Hasegawa, Y.; Hasegawa, T. A novel KAL1 mutation is associated with combined pituitary hormone deficiency. Hum. Genome Var. 2014, 1, 14011. [Google Scholar] [CrossRef]
- Tata, B.; Huijbregts, L.; Jacquier, S.; Csaba, Z.; Genin, E.; Meyer, V.; Leka, S.; Dupont, J.; Charles, P.; Chevenne, D.; et al. Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol. 2014, 12, e1001952. [Google Scholar] [CrossRef]
- Thakur, M.; Taha, D.; Misra, V.K. A Case of Congenital Hypopituitarism Associated With a 1p31 Microdeletion: A Possible Role for. J. Endocr. Soc. 2017, 1, 278–282. [Google Scholar] [CrossRef][Green Version]
- Tommiska, J.; Känsäkoski, J.; Skibsbye, L.; Vaaralahti, K.; Liu, X.; Lodge, E.J.; Tang, C.; Yuan, L.; Fagerholm, R.; Kanters, J.K.; et al. Two missense mutations in KCNQ1 cause pituitary hormone deficiency and maternally inherited gingival fibromatosis. Nat. Commun. 2017, 8, 1289. [Google Scholar] [CrossRef]
- Webb, E.A.; AlMutair, A.; Kelberman, D.; Bacchelli, C.; Chanudet, E.; Lescai, F.; Andoniadou, C.L.; Banyan, A.; Alsawaid, A.; Alrifai, M.T.; et al. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain 2013, 136, 3096–3105. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]
- Lerario, A.M.; Mohan, D.R.; Montenegro, L.R.; Funari, M.F.A.; Nishi, M.Y.; Narcizo, A.M.; Benedetti, A.F.F.; Oba-Shinjo, S.M.; Vitorino, A.J.; Santos, R.A.S.X.; et al. SELAdb: A database of exonic variants in a Brazilian population referred to a quaternary medical center in São Paulo. Clinics 2020, 75, e1913. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M. Functional correlation between cell adhesive properties and some cell surface proteins. J. Cell Biol. 1977, 75, 464–474. [Google Scholar] [CrossRef]
- Phillips, J.A.; Hjelle, B.L.; Seeburg, P.H.; Zachmann, M. Molecular basis for familial isolated growth hormone deficiency. Proc. Natl. Acad. Sci. USA 1981, 78, 6372–6375. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M. Genetic causes of isolated and combined pituitary hormone deficiency. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Mullis, P.E. Genetics of growth hormone deficiency. Endocrinol. Metab. Clin. N. Am. 2007, 36, 17–36. [Google Scholar] [CrossRef]
- Alatzoglou, K.S.; Dattani, M.T. Genetic forms of hypopituitarism and their manifestation in the neonatal period. Early Hum. Dev. 2009, 85, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Jelsig, A.M.; Diness, B.R.; Kreiborg, S.; Main, K.M.; Larsen, V.A.; Hove, H. A complex phenotype in a family with a pathogenic SOX3 missense variant. Eur. J. Med. Genet. 2018, 61, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chang, G.; Cheng, Q.; Yao, R.; Li, J.; Xu, Y.; Li, G.; Ding, Y.; Qing, Y.; Li, N.; et al. Increased transactivation and impaired repression of β-catenin-mediated transcription associated with a novel SOX3 missense mutation in an X-linked hypopituitarism pedigree with modest growth failure. Mol. Cell Endocrinol. 2018, 478, 133–140. [Google Scholar] [CrossRef]
- Alatzoglou, K.S.; Kelberman, D.; Cowell, C.T.; Palmer, R.; Arnhold, I.J.; Melo, M.E.; Schnabel, D.; Grueters, A.; Dattani, M.T. Increased transactivation associated with SOX3 polyalanine tract deletion in a patient with hypopituitarism. J. Clin. Endocrinol. Metab. 2011, 96, E685–E690. [Google Scholar] [CrossRef]
- Chen, C.P.; Chern, S.R.; Du, S.H.; Wang, W. Molecular diagnosis of a novel heterozygous 268C-->T (R90C) mutation in TGIF gene in a fetus with holoprosencephaly and premaxillary agenesis. Prenat. Diagn. 2002, 22, 5–7. [Google Scholar] [CrossRef]
- Traggiai, C.; Stanhope, R. Endocrinopathies associated with midline cerebral and cranial malformations. J. Pediatr. 2002, 140, 252–255. [Google Scholar] [CrossRef]
- El-Jaick, K.B.; Powers, S.E.; Bartholin, L.; Myers, K.R.; Hahn, J.; Orioli, I.M.; Ouspenskaia, M.; Lacbawan, F.; Roessler, E.; Wotton, D.; et al. Functional analysis of mutations in TGIF associated with holoprosencephaly. Mol. Genet. Metab. 2007, 90, 97–111. [Google Scholar] [CrossRef]
- Gripp, K.W.; Wotton, D.; Edwards, M.C.; Roessler, E.; Ades, L.; Meinecke, P.; Richieri-Costa, A.; Zackai, E.H.; Massagué, J.; Muenke, M.; et al. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat. Genet. 2000, 25, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Melhuish, T.A.; Wotton, D. The interaction of the carboxyl terminus-binding protein with the Smad corepressor TGIF is disrupted by a holoprosencephaly mutation in TGIF. J. Biol. Chem. 2000, 275, 39762–39766. [Google Scholar] [CrossRef] [PubMed]
- Tatsi, C.; Sertedaki, A.; Voutetakis, A.; Valavani, E.; Magiakou, M.A.; Kanaka-Gantenbein, C.; Chrousos, G.P.; Dacou-Voutetakis, C. Pituitary stalk interruption syndrome and isolated pituitary hypoplasia may be caused by mutations in holoprosencephaly-related genes. J. Clin. Endocrinol. Metab. 2013, 98, E779–E784. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age at Testing in Years | Initial Height SDS | Puberty I/S (Years) | Final Height SDS | Target Height SDS | GH Peak μg/L | Cortisol Peak n·mol/L (NR > 550) | FT4 p·mol/L (NR) | TSH mU/L (NR) | IGF1 ng/mL (NR) | IGFBP3 mg/L (NR) | PRL mU/L (NR) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 9.5 | −7.65 | S (14) | −0.63 | −0.63 | <0.25 | NA | NA | 6.0 (0.5–4.4) | NA | NA | 54 |
| 2 | 6 | −4.67 | S (13) | −3.2 | −0.7 | 0.9 | 552 | 0.97 (0.7–1.5) | 2.37 (0.5–4.4) | 200 (227–964) | 4.2 (3.3–5.7) | 340 (<450) |
| 3 | 0.66 | −4.8 | - | Still growing | +0.55 | 0.15 * | 39 | ** | 6.3 (0–20) | 25 (48-313) | NA | 278 (57–717) |
| 4 | 4.9 | −4.55 | I (15) | −0.93 | 0.34 | 0.4 | NA | *** | 3.11 (0.5–4.2) | <18 (25–68) | 0.4 (1.5–3.4) | 42.5 (42-170) |
| Patient | M/F | Gene | Allelic Variant | Inheritance | Hormone Deficiencies | MRI (CA) |
|---|---|---|---|---|---|---|
| 1 | M | GH1 | p.Phe57Leufs*43 | AR | IGHD HyperHypogon | TPP normal AP 7.5 year |
| 2 | M | SOX3 | p.Met304Ile | X-linked | IGHD | EPP AP aplasia 8 year |
| 3 | F | TGIF1 | p.Arg90Cys | AD–IC | GH, TSH, ACTH, PRL and ADH | HPE 40 days |
| 4 | M | TGIF1 | p.Ser28Pro | AD–IC | GH, TSH, ACTH, LH/FSH, PRL | EPP AP aplasia 5.1 year |
| Gene | Variant | OMIM/Genecards | gnomAD | ABraOM | SELAdb | ACMG | CADD | REVEL |
|---|---|---|---|---|---|---|---|---|
| GH1 | NM_000515.5:c.171delT; p.Phe57Leufs*43 (chr17:61995706: delA) | Never related to hypopituitarism | Absent | Absent | Absent | Pathogenic | 22.4 | No data |
| SOX3 | NM_005634.3:c.912G>A;p.Met304Ile (chrX:139586314: C>T) | Never related to hypopituitarism | Absent | Absent | Absent | VUS | 23.4 | 0.670 |
| TGIF1 | NM_173208.1 c.268C>T: p.Arg90Cys (chr18:3457387: C>T) | Never related to hypopituitarism | Absent | Absent | Absent | Likely pathogenic | 28.8 | 0.9729 |
| TGIF1 | NM_173208.1 c.82T>C; p.Ser28Pro (chr18:3456417: T>C) | Never related to hypopituitarism | Absent | Absent | Absent | VUS | 22.8 | 0.279 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakaguma, M.; Ferreira, N.G.B.P.; Benedetti, A.F.F.; Madi, M.C.; Silva, J.M.; Li, J.Z.; Ma, Q.; Bilge Ozel, A.; Fang, Q.; Narcizo, A.d.M.; et al. Allelic Variants in Established Hypopituitarism Genes Expand Our Knowledge of the Phenotypic Spectrum. Genes 2021, 12, 1128. https://doi.org/10.3390/genes12081128
Nakaguma M, Ferreira NGBP, Benedetti AFF, Madi MC, Silva JM, Li JZ, Ma Q, Bilge Ozel A, Fang Q, Narcizo AdM, et al. Allelic Variants in Established Hypopituitarism Genes Expand Our Knowledge of the Phenotypic Spectrum. Genes. 2021; 12(8):1128. https://doi.org/10.3390/genes12081128
Chicago/Turabian StyleNakaguma, Marilena, Nathalia Garcia Bianchi Pereira Ferreira, Anna Flavia Figueredo Benedetti, Mariana Cotarelli Madi, Juliana Moreira Silva, Jun Z. Li, Qianyi Ma, Ayse Bilge Ozel, Qing Fang, Amanda de Moraes Narcizo, and et al. 2021. "Allelic Variants in Established Hypopituitarism Genes Expand Our Knowledge of the Phenotypic Spectrum" Genes 12, no. 8: 1128. https://doi.org/10.3390/genes12081128
APA StyleNakaguma, M., Ferreira, N. G. B. P., Benedetti, A. F. F., Madi, M. C., Silva, J. M., Li, J. Z., Ma, Q., Bilge Ozel, A., Fang, Q., Narcizo, A. d. M., Cardoso, L. C., Montenegro, L. R., Funari, M. F. d. A., Nishi, M. Y., Arnhold, I. J. P., Jorge, A. A. d. L., Mendonca, B. B. d., Camper, S. A., & Carvalho, L. R. (2021). Allelic Variants in Established Hypopituitarism Genes Expand Our Knowledge of the Phenotypic Spectrum. Genes, 12(8), 1128. https://doi.org/10.3390/genes12081128