
Store-Independent Calcium Entry and Related Signaling Pathways in Breast Cancer


{kind=link}
Abstract
1. Introduction
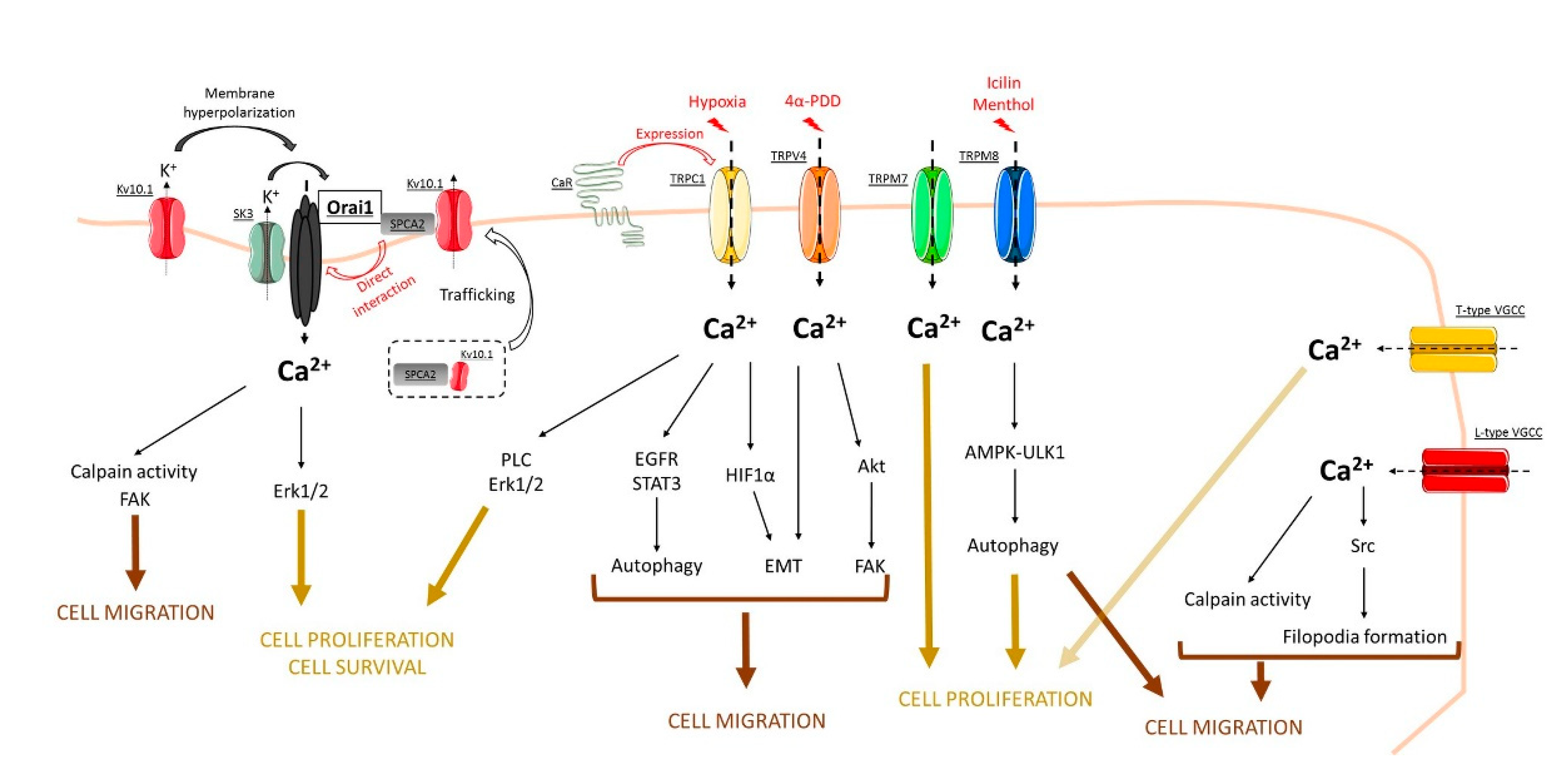
2. Orai Channels
3. TRP Channels
4. Voltage-Gated Calcium Channels
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid; |
| AMPK-ULK1 | AMP-activated protein kinase–Unc-51 like autophagy activating kinase 1; |
| BC | Breast cancer; |
| Ca2+ | Calcium; |
| CaR | Calcium-sensing receptor; |
| C-ter | carboxyl terminus; |
| EGFR | Epidermal growth factor; |
| EMT | Epithelial-mesenchymal transition; |
| ER+ | Estrogen receptor positive; |
| ER− | Estrogen receptor negative; |
| Erk1/2 | Extracellular signal-regulated kinases 1 and 2; |
| FAK | Focal adhesion kinase; |
| HIF1α | Hypoxia-inducible factor 1 α; |
| Kv10.1 | Voltage-gated potassium channel; |
| LTC4 | AA metabolite leukotriene C4 (LTC4); |
| miRNA | Micro-ribonucleic acid; |
| N-ter | Amino terminus; |
| PLC | Phospholipase C; |
| SICE | Store-independent calcium entry; |
| SK3 | Small conductance calcium-activated potassium channel; |
| SOC | Store-operated channels; |
| SOCE | Store-operated calcium entry; |
| SOX4 | SRY-Box transcription factor 4; |
| SPCA2 | Secretory pathway calcium-ATPase; |
| Src | Proto-oncogene tyrosine kinase Src; |
| STAT3 | Signal transducers and activators of transcription 3; |
| STIM | Stromal interaction molecule; |
| TRP | Transient receptor potential; |
| TRPC | Transient receptor potential canonical; |
| TRPM | Transient receptor potential melastatin; |
| TRPV | Transient receptor potential vanilloid; |
| VGCC | Voltage-gated calcium channel; |
| 4α-PDD | 4-α-phorbol-12,13-didecanoate. |
References
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skrryma, R.; Shuuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef]
- Tajada, S.; Villalobos, C. Calcium Permeable Channels in Cancer Hallmarks. Front. Pharmacol. 2020, 11, 968. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H.; Michalak, M.; Verkhratsky, A. Calcium signalling: Past, present and future. Cell Calcium 2005, 38, 161–169. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef]
- So, C.L.; Saunus, J.M.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium signalling and breast cancer. Semin. Cell Dev. Biol. 2019, 94, 74–83. [Google Scholar] [CrossRef]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Store-Operated Ca(2+) Entry in Breast Cancer Cells: Remodeling and Functional Role. Int. J. Mol. Sci. 2018, 19, 4053. [Google Scholar] [CrossRef]
- Zhang, X.; Gueguinou, M.; Trebak, M. Store-Independent Orai Channels Regulated by STIM. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; CRC Press: Boca Raton, FL, USA, 2018; pp. 197–214. [Google Scholar]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Phan, N.N.; Wang, C.Y.; Chen, C.F.; Sun, Z.; Lai, M.D.; Lin, Y.C. Voltage-gated calcium channels: Novel targets for cancer therapy. Oncol. Lett. 2017, 14, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Deliot, N. Constantin, B. Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochim. Biophys. Acta 2015, 1848, 2512–2522. [Google Scholar] [CrossRef]
- Mignen, O.; Constantin, B.; Potier-Cartereau, M.; Penna, A.; Gautier, M.; Gueguinou, M.; Renaudineau, Y.; Shoji, K.F.; Felix, R.; Bayet, E.; et al. Constitutive calcium entry and cancer: Updated views and insights. Eur. Biophys. J. EBJ 2017, 46, 395–413. [Google Scholar] [CrossRef]
- Chen, Y.F.; Lin, P.C.; Yeh, Y.M.; Chen, L.H.; Shen, M.R. Store-Operated Ca(2+) Entry in Tumor Progression: From Molecular Mechanisms to Clinical Implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta 2013, 1833, 752–760. [Google Scholar] [CrossRef]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Milevskiy, M.J.G.; Chalmers, S.B.; Yapa, K.; Robitaille, M.; Henry, C.; Baillie, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses. Cancers 2019, 11, 208. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.; Tanwar, J.; Motiani, R.K. Regulation of proto-oncogene Orai3 by miR18a/b and miR34a. Cell Calcium 2018, 75, 101–111. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.M.; Hack, A.; Reinhardt, T.A.; Rao, R. SPCA2 regulates Orai1 trafficking and store independent Ca2+ entry in a model of lactation. PLoS ONE 2013, 8, e67348. [Google Scholar] [CrossRef]
- Peretti, M.; Badaoui, M.; Girault, A.; van Gulick, L.; Mabille, M.P.; Tebbakha, R.; Sevestre, H.; Morjani, H.; Ouadid-Ahidouch, H. Original association of ion transporters mediates the ECM-induced breast cancer cell survival: Kv10.1-Orai1-SPCA2 partnership. Sci. Rep. 2019, 9, 1175. [Google Scholar] [CrossRef] [PubMed]
- Girault, A.; Peretti, M.; Badaoui, M.; Hemon, A.; Morjani, H.; Ouadid-Ahidouch, H. The N and C-termini of SPCA2 regulate differently Kv10.1 function: Role in the collagen 1-induced breast cancer cell survival. Am. J. Cancer Res. 2021, 11, 251–263. [Google Scholar]
- Badaoui, M.; Mimsy-Julienne, C.; Saby, C.; van Gulick, L.; Peretti, M.; Jeannesson, P.; Morjani, H.; Ouadid-Ahidouch, H. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10.1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget 2018, 9, 24653–24671. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Hanley, P.; Fabian, A.; Stock, C. Potassium channels keep mobile cells on the go. Physiology 2008, 23, 212–220. [Google Scholar] [CrossRef][Green Version]
- Hammadi, M.; Chopin, V.; Matifat, F.; Dhennin-Duthille, I.; Chasseraud, M.; Sevestre, H.; Ouadid-Ahidouch, H. Human ether a-gogo K(+) channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J. Cell. Physiol. 2012, 227, 3837–3846. [Google Scholar] [CrossRef]
- Chantome, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Gueguinou, M.; Pages, J.C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef]
- Gueguinou, M.; Crottes, D.; Chantome, A.; Rapetti-Mauss, R.; Potier-Cartereau, M.; Clarysse, L.; Girault, A.; Fourbon, Y.; Jezequel, P.; Guerin-Charbonnel, C.; et al. The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca(2+) homeostasis. Oncogene 2017, 36, 3640–3647. [Google Scholar] [CrossRef]
- Chodon, D.; Guilbert, A.; Dhennin-Duthille, I.; Gautier, M.; Telliez, M.S.; Sevestre, H.; Ouadid-Ahidouch, H. Estrogen regulation of TRPM8 expression in breast cancer cells. BMC Cancer 2010, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2011, 28, 813–822. [Google Scholar] [CrossRef] [PubMed]
- El Hiani, Y.; Ahidouch, A.; Roudbaraki, M.; Guenin, S.; Brule, G.; Ouadid-Ahidouch, H. Calcium-sensing receptor stimulation induces nonselective cation channel activation in breast cancer cells. J. Membr. Biol. 2006, 211, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Guilbert, A.; Dhennin-Duthille, I.; Hiani, Y.E.; Haren, N.; Khorsi, H.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Expression of TRPC6 channels in human epithelial breast cancer cells. BMC Cancer 2008, 8, 125. [Google Scholar] [CrossRef]
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Haren, N.; Sevestre, H.; Ouadid-Ahidouch, H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am. J. Physiol. Cell Physiol. 2009, 297, C493–C502. [Google Scholar] [CrossRef]
- Ouadid-Ahidouch, H.; Dhennin-Duthille, I.; Gautier, M.; Sevestre, H.; Ahidouch, A. TRP calcium channel and breast cancer: Expression, role and correlation with clinical parameters. Bull. Cancer 2012, 99, 655–664. [Google Scholar] [CrossRef]
- Aydar, E.; Yeo, S.; Djamgoz, M.; Palmer, C. Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: A potential target for breast cancer diagnosis and therapy. Cancer Cell Int. 2009, 9, 23. [Google Scholar] [CrossRef]
- Lee, W.H.; Choong, L.Y.; Jin, T.H.; Mon, N.N.; Chong, S.; Liew, C.S.; Putti, T.; Lu, S.Y.; Harteneck, C.; Lim, Y.P. TRPV4 plays a role in breast cancer cell migration via Ca(2+)-dependent activation of AKT and downregulation of E-cadherin cell cortex protein. Oncogenesis 2017, 6, e338. [Google Scholar] [CrossRef]
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Rybarczyk, P.; Sahni, J.; Sevestre, H.; Scharenberg, A.M.; Ouadid-Ahidouch, H. Transient receptor potential melastatin 7 is involved in oestrogen receptor-negative metastatic breast cancer cells migration through its kinase domain. Eur. J. Cancer 2013, 49, 3694–3707. [Google Scholar] [CrossRef]
- Meng, X.; Cai, C.; Wu, J.; Cai, S.; Ye, C.; Chen, H.; Yang, Z.; Zeng, H.; Shen, Q.; Zou, F. TRPM7 mediates breast cancer cell migration and invasion through the MAPK pathway. Cancer Lett. 2013, 333, 96–102. [Google Scholar] [CrossRef]
- El Hiani, Y.; Ahidouch, A.; Lehen’kyi, V.; Hague, F.; Gouilleux, F.; Mentaverri, R.; Kamel, S.; Lassoued, K.; Brule, G.; Ouadid-Ahidouch, H. Extracellular signal-regulated kinases 1 and 2 and TRPC1 channels are required for calcium-sensing receptor-stimulated MCF-7 breast cancer cell proliferation. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef]
- Sanders, J.L.; Chattopadhyay, N.; Kifor, O.; Yamaguchi, T.; Butters, R.R.; Brown, E.M. Extracellular calcium-sensing receptor expression and its potential role in regulating parathyroid hormone-related peptide secretion in human breast cancer cell lines. Endocrinology 2000, 141, 4357–4364. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Milevskiy, M.J.G.; Kaemmerer, E.; Turner, D.; Yapa, K.; Brown, M.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells. J. Cell Sci. 2017, 130, 2292–2305. [Google Scholar] [CrossRef]
- Azimi, I.; Robitaille, M.; Armitage, K.; So, C.L.; Milevskiy, M.J.G.; Northwood, K.; Lim, H.F.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Activation of the Ion Channel TRPV4 Induces Epithelial to Mesenchymal Transition in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9417. [Google Scholar] [CrossRef]
- Middelbeek, J.; Kuipers, A.J.; Henneman, L.; Visser, D.; Eidhof, I.; van Horssen, R.; Wieringa, B.; Canisius, S.V.; Zwart, W.; Wessels, L.F.; et al. TRPM7 is required for breast tumor cell metastasis. Cancer Res. 2012, 72, 4250–4261. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, A.J.; Middelbeek, J.; Vrenken, K.; Perez-Gonzalez, C.; Poelmans, G.; Klarenbeek, J.; Jalink, K.; Trepat, X.; van Leeuwen, F.N. TRPM7 controls mesenchymal features of breast cancer cells by tensional regulation of SOX4. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, S.; Jia, Z.; Zhao, W.; Zhou, C.; Zhang, R.; Ali, D.W.; Michalak, M.; Chen, X.Z.; Tang, J. Transient Receptor Potential Melastatin 8 (TRPM8) Channel Regulates Proliferation and Migration of Breast Cancer Cells by Activating the AMPK-ULK1 Pathway to Enhance Basal Autophagy. Front. Oncol. 2020, 10, 573127. [Google Scholar] [CrossRef]
- Roger, S.; Besson, P.; le Guennec, J.Y. Involvement of a novel fast inward sodium current in the invasion capacity of a breast cancer cell line. Biochim. Biophys. Acta 2003, 1616, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Wonderlin, W.F.; Woodfork, K.A.; Strobl, J.S. Changes in membrane potential during the progression of MCF-7 human mammary tumor cells through the cell cycle. J. Cell. Physiol. 1995, 165, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; le Bourhis, X.; Roudbaraki, M.; Toillon, R.A.; Delcourt, P.; Prevarskaya, N. Changes in the K+ current-density of MCF-7 cells during progression through the cell cycle: Possible involvement of a h-ether.a-gogo K+ channel. Recept. Channels 2001, 7, 345–356. [Google Scholar]
- Barcelo, C.; Siso, P.; Maiques, O.; de la Rosa, I.; Marti, R.M.; Macia, A. T-Type Calcium Channels: A Potential Novel Target in Melanoma. Cancers 2020, 12, 391. [Google Scholar] [CrossRef]
- Ohkubo, T.; Yamazaki, J. T-type voltage-activated calcium channel Cav3.1, but not Cav3.2, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 267–275. [Google Scholar] [CrossRef]
- Wang, C.Y.; Lai, M.D.; Phan, N.N.; Sun, Z.; Lin, Y.C. Meta-Analysis of Public Microarray Datasets Reveals Voltage-Gated Calcium Gene Signatures in Clinical Cancer Patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.; Peres, C.G.; Vaz, C.V.; Gomes, I.M.; Figueira, M.I.; Cairrao, E.; Verde, I.; Maia, C.J.; Socorro, S. 5alpha-Dihydrotestosterone regulates the expression of L-type calcium channels and calcium-binding protein regucalcin in human breast cancer cells with suppression of cell growth. Med Oncol. 2015, 32, 228. [Google Scholar] [CrossRef] [PubMed]
- Squecco, R.; Tani, A.; Zecchi-Orlandini, S.; Formigli, L.; Francini, F. Melatonin affects voltage-dependent calcium and potassium currents in MCF-7 cell line cultured either in growth or differentiation medium. Eur. J. Pharmacol. 2015, 758, 40–52. [Google Scholar] [CrossRef]
- Kanwar, N.; Carmine-Simmen, K.; Nair, R.; Wang, C.; Moghadas-Jafari, S.; Blaser, H.; Tran-Thanh, D.; Wang, D.; Wang, P.; Wang, J.; et al. Amplification of a calcium channel subunit CACNG4 increases breast cancer metastasis. EBioMedicine 2020, 52, 102646. [Google Scholar] [CrossRef]
- Jacquemet, G.; Baghirov, H.; Georgiadou, M.; Sihto, H.; Peuhu, E.; Cettour-Janet, P.; He, T.; Perala, M.; Kronqvist, P.; Joensuu, H.; et al. L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat. Commun. 2016, 7, 13297. [Google Scholar] [CrossRef]
- Bertolesi, G.E.; Shi, C.; Elbaum, L.; Jollimore, C.; Rozenberg, G.; Barnes, S.; Kelly, M.E. The Ca(2+) channel antagonists mibefradil and pimozide inhibit cell growth via different cytotoxic mechanisms. Mol. Pharmacol. 2002, 62, 210–219. [Google Scholar] [CrossRef]
- Taylor, J.T.; Huang, L.; Pottle, J.E.; Liu, K.; Yang, Y.; Zeng, X.; Keyser, B.M.; Agrawal, K.C.; Hansen, J.B.; Li, M. Selective blockade of T-type Ca2+ channels suppresses human breast cancer cell proliferation. Cancer Lett. 2008, 267, 116–124. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamlali, M.; Rodat-Despoix, L.; Ouadid-Ahidouch, H. Store-Independent Calcium Entry and Related Signaling Pathways in Breast Cancer. Genes 2021, 12, 994. https://doi.org/10.3390/genes12070994
Chamlali M, Rodat-Despoix L, Ouadid-Ahidouch H. Store-Independent Calcium Entry and Related Signaling Pathways in Breast Cancer. Genes. 2021; 12(7):994. https://doi.org/10.3390/genes12070994
Chicago/Turabian StyleChamlali, Mohamed, Lise Rodat-Despoix, and Halima Ouadid-Ahidouch. 2021. "Store-Independent Calcium Entry and Related Signaling Pathways in Breast Cancer" Genes 12, no. 7: 994. https://doi.org/10.3390/genes12070994
APA StyleChamlali, M., Rodat-Despoix, L., & Ouadid-Ahidouch, H. (2021). Store-Independent Calcium Entry and Related Signaling Pathways in Breast Cancer. Genes, 12(7), 994. https://doi.org/10.3390/genes12070994