Role of Sigma-1 Receptor in Calcium Modulation: Possible Involvement in Cancer



{kind=link}
Abstract
1. Introduction
2. Ca2+ Homeostasis Regulation in the ER
3. The Mitochondria Associated Membranes
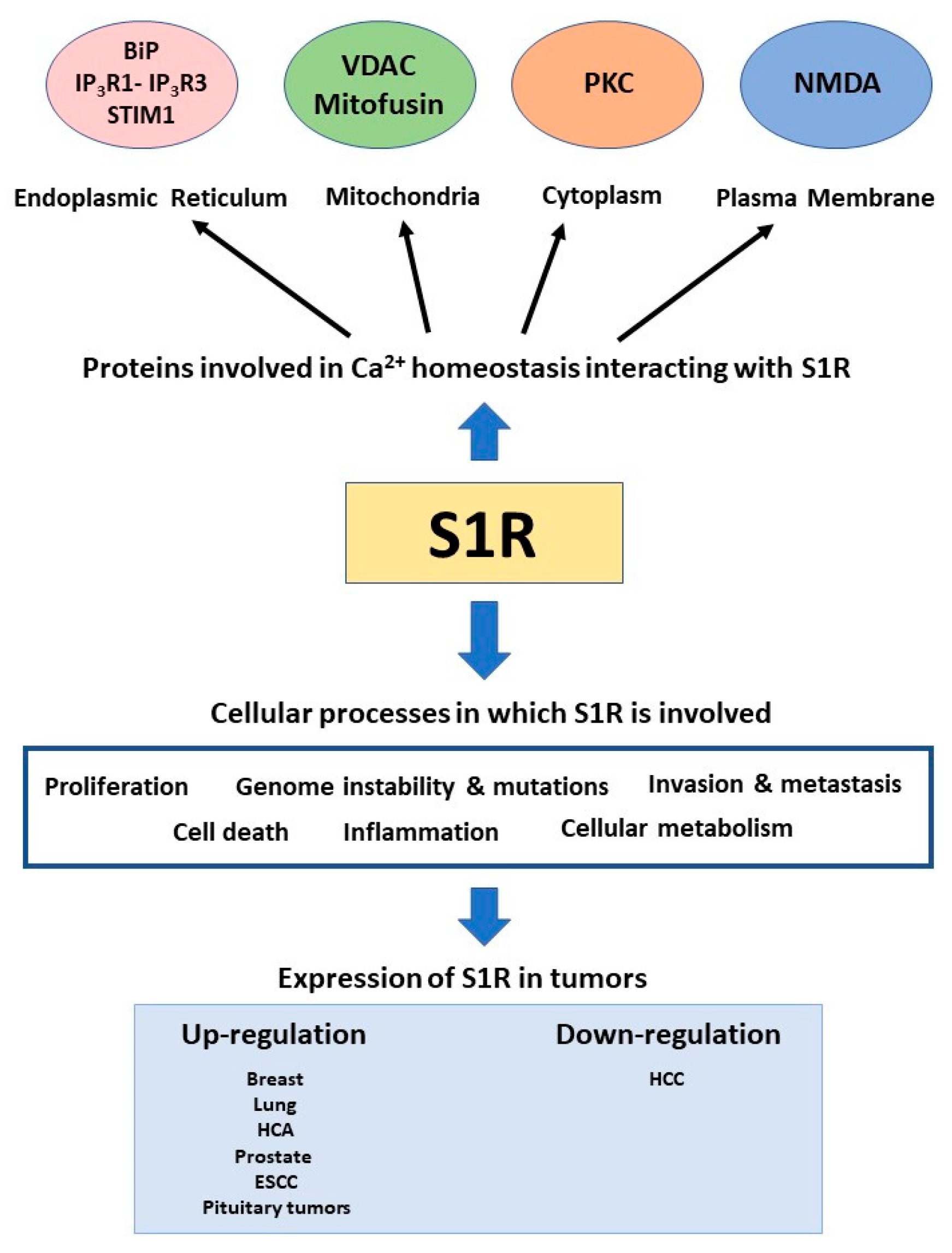
4. The Sigma-1 Receptor
5. Sigma-1 Receptor in Cancer
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The Versatility and Universality of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca(2+) Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca(2+) Fluxes and Cancer. Mol. Cell. 2020, 78, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The Calcium-Cancer Signalling Nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-Associated Membranes: Composition, Molecular Mechanisms, and Physiopathological Implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Danese, A.; Marchi, S.; Vitto, V.A.M.; Modesti, L.; Leo, S.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Cancer-Related Increases and Decreases in Calcium Signaling at the Endoplasmic Reticulum-Mitochondria Interface (MAMs). In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Tsai, S.-Y.; Hayashi, T.; Mori, T.; Su, T.-P. Sigma-1 Receptor Chaperones and Diseases. Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 184–189. [Google Scholar] [CrossRef]
- Su, T.-P.; Su, T.-C.; Nakamura, Y.; Tsai, S.-Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2+) Homeostasis and Endoplasmic Reticulum (ER) Stress: An Integrated View of Calcium Signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihan, P.; Hetz, C. Calcium Signaling at the Endoplasmic Reticulum: Fine-Tuning Stress Responses. Cell Calcium 2018, 70, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J.; Pozzan, T. The Endoplasmic Reticulum Ca2+ Store: A View from the Lumen. Trends Biochem. Sci. 1998, 23, 10–14. [Google Scholar] [CrossRef]
- Coe, H.; Michalak, M. Calcium Binding Chaperones of the Endoplasmic Reticulum. Gen. Physiol. Biophys. 2009, 28, F96–F103. [Google Scholar]
- Primeau, J.O.; Armanious, G.P.; Fisher, M.E.; Young, H.S. The SarcoEndoplasmic Reticulum Calcium ATPase. Subcell. Biochem. 2018, 87, 229–258. [Google Scholar] [PubMed]
- Parys, J.B.; Vervliet, T. New Insights in the IP(3) Receptor and Its Regulation. Adv. Exp. Med. Biol. 2020, 1131, 243–270. [Google Scholar]
- Lanner, J.T. Ryanodine Receptor Physiology and Its Role in Disease. Adv. Exp. Med. Biol. 2012, 740, 217–234. [Google Scholar]
- Camello, C.; Lomax, R.; Petersen, O.H.; Tepikin, A. V Calcium Leak from Intracellular Stores—The Enigma of Calcium Signalling. Cell Calcium 2002, 32, 355–361. [Google Scholar] [CrossRef]
- Vanderheyden, V.; Devogelaere, B.; Missiaen, L.; De Smedt, H.; Bultynck, G.; Parys, J.B. Regulation of Inositol 1,4,5-Trisphosphate-Induced Ca2+ Release by Reversible Phosphorylation and Dephosphorylation. Biochim. Biophys. Acta 2009, 1793, 959–970. [Google Scholar] [CrossRef]
- Parys, J.B.; De Smedt, H. Inositol 1,4,5-Trisphosphate and Its Receptors. Adv. Exp. Med. Biol. 2012, 740, 255–279. [Google Scholar]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca(2+) Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A Forty-Kilodalton Protein of the Inner Membrane is the Mitochondrial Calcium Uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Bruni, R.; Kloss, B.; Assur, Z.; Kloppmann, E.; Rost, B.; Hendrickson, W.A.; Liu, Q. Structural Basis for a PH-Sensitive Calcium Leak across Membranes. Science 2014, 344, 1131–1135. [Google Scholar] [CrossRef]
- Lisak, D.A.; Schacht, T.; Enders, V.; Habicht, J.; Kiviluoto, S.; Schneider, J.; Henke, N.; Bultynck, G.; Methner, A. The Transmembrane Bax Inhibitor Motif (TMBIM) Containing Protein Family: Tissue Expression, Intracellular Localization and Effects on the ER CA2+-Filling State. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2104–2114. [Google Scholar] [CrossRef] [PubMed]
- Lomax, R.B.; Camello, C.; Van Coppenolle, F.; Petersen, O.H.; Tepikin, A. V Basal and Physiological Ca(2+) Leak from the Endoplasmic Reticulum of Pancreatic Acinar Cells. Second Messenger-Activated Channels and Translocons. J. Biol. Chem. 2002, 277, 26479–26485. [Google Scholar] [CrossRef] [PubMed]
- Van Coppenolle, F.; Vanden Abeele, F.; Slomianny, C.; Flourakis, M.; Hesketh, J.; Dewailly, E.; Prevarskaya, N. Ribosome-Translocon Complex Mediates Calcium Leakage from Endoplasmic Reticulum Stores. J. Cell Sci. 2004, 117, 4135–4142. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; Liu, X.; Sharma, A.; Hegde, R.S.; Ambudkar, I.S. Intracellular Ca(2+) Release via the ER Translocon Activates Store-Operated Calcium Entry. Pflugers Arch. 2007, 453, 797–808. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef]
- Zhang, A.; Williamson, C.D.; Wong, D.S.; Bullough, M.D.; Brown, K.J.; Hathout, Y.; Colberg-Poley, A.M. Quantitative Proteomic Analyses of Human Cytomegalovirus-Induced Restructuring of Endoplasmic Reticulum-Mitochondrial Contacts at Late Times of Infection. Mol. Cell. Proteom. 2011, 10, M111.009936. [Google Scholar] [CrossRef]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-Depth Proteomic Analysis of Mammalian Mitochondria-Associated Membranes (MAM). J. Proteom. 2013, 79, 219–230. [Google Scholar] [CrossRef]
- Sala-Vila, A.; Navarro-Lérida, I.; Sánchez-Alvarez, M.; Bosch, M.; Calvo, C.; López, J.A.; Calvo, E.; Ferguson, C.; Giacomello, M.; Serafini, A.; et al. Interplay between Hepatic Mitochondria-Associated Membranes, Lipid Metabolism and Caveolin-1 in Mice. Sci. Rep. 2016, 6, 27351. [Google Scholar] [CrossRef]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-Associated Membranes (MAMs) and Inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-Associated ER Membranes—The Origin Site of Autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef]
- Vecellio Reane, D.; Rizzuto, R.; Raffaello, A. The ER-Mitochondria Tether at the Hub of Ca2+ Signaling. Curr. Opin. Physiol. 2020, 17, 261–268. [Google Scholar] [CrossRef]
- Wacquier, B.; Combettes, L.; Van Nhieu, G.T.; Dupont, G. Interplay Between Intracellular Ca(2+) Oscillations and Ca(2+)-Stimulated Mitochondrial Metabolism. Sci. Rep. 2016, 6, 19316. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial Calcium Signalling and Cell Death: Approaches for Assessing the Role of Mitochondrial Ca2+ Uptake in Apoptosis. Cell Calcium. 2006, 40, 553–560. [Google Scholar] [CrossRef]
- Higo, T.; Hattori, M.; Nakamura, T.; Natsume, T.; Michikawa, T.; Mikoshiba, K. Subtype-Specific and ER Lumenal Environment-Dependent Regulation of Inositol 1,4,5-Trisphosphate Receptor Type 1 by ERp44. Cell 2005, 120, 85–98. [Google Scholar] [CrossRef]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1α Regulates Ca(2+) Fluxes at the Endoplasmic Reticulum-Mitochondria Interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef]
- Zhou, Z.; Torres, M.; Sha, H.; Halbrook, C.J.; Van den Bergh, F.; Reinert, R.B.; Yamada, T.; Wang, S.; Luo, Y.; Hunter, A.H.; et al. Endoplasmic Reticulum–Associated Degradation Regulates Mitochondrial Dynamics in Brown Adipocytes. Science 2020, 368, 54–60. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, X.; Xu, J.; Shang, W.; Tong, C. A Novel Fluorescent Reporter Detects Plastic Remodeling of Mitochondria-ER Contact Sites. J. Cell Sci. 2018, 131, jcs208686. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. SPLICS: A Split Green Fluorescent Protein-Based Contact Site Sensor for Narrow and Wide Heterotypic Organelle Juxtaposition. Cell Death Differ. 2018, 25, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-Mitochondrial Coupling Promotes Mitochondrial Respiration and Bioenergetics during Early Phases of ER Stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; López-Crisosto, C.; Parra, V.; Rodriguez-Peña, M.; Rothermel, B.A.; Quest, A.F.G.; Lavandero, S. MTORC1 Inhibitor Rapamycin and ER Stressor Tunicamycin Induce Differential Patterns of ER-Mitochondria Coupling. Sci. Rep. 2016, 6, 36394. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK Is Required at the ER-Mitochondrial Contact Sites to Convey Apoptosis after ROS-Based ER Stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 Modulates the UPR and Mitochondrial Function via Repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.-P. Sigma-1 Receptor Chaperone at the ER-Mitochondrion Interface Mediates the Mitochondrion-ER-Nucleus Signaling for Cellular Survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Jana, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernandez, E.; Sassano, M.L.; et al. Non-Canonical Function of IRE1alpha Determines Mitochondria-Associated Endoplasmic Reticulum Composition to Control Calcium Transfer and Bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Martinvalet, D. The Role of the Mitochondria and the Endoplasmic Reticulum Contact Sites in the Development of the Immune Responses. Cell Death Dis. 2018, 9, 336. [Google Scholar] [CrossRef]
- Rieusset, J. The Role of Endoplasmic Reticulum-Mitochondria Contact Sites in the Control of Glucose Homeostasis: An Update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, X. Endoplasmic Reticulum-Mitochondria Tethering in Neurodegenerative Diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The Effects of Morphine- and Nalorphine- like Drugs in the Nondependent and Morphine-Dependent Chronic Spinal Dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [PubMed]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, Molecular Cloning, and Expression of the Mammalian Sigma1-Binding Site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Ligands: Potential in the Treatment of Neuropsychiatric Disorders. CNS Drugs 2004, 18, 269–284. [Google Scholar] [CrossRef]
- Chu, U.B.; Ramachandran, S.; Hajipour, A.R.; Ruoho, A.E. Photoaffinity Labeling of the Sigma-1 Receptor with N-[3-(4-Nitrophenyl)Propyl]-N-Dodecylamine: Evidence of Receptor Dimers. Biochemistry 2013, 52, 859–868. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal Structure of the Human Σ1 Receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- Ortega-Roldan, J.L.; Ossa, F.; Amin, N.T.; Schnell, J.R. Solution NMR Studies Reveal the Location of the Second Transmembrane Domain of the Human Sigma-1 Receptor. FEBS Lett. 2015, 589, 659–665. [Google Scholar] [CrossRef]
- Penke, B.; Fulop, L.; Szucs, M.; Frecska, E. The Role of Sigma-1 Receptor, an Intracellular Chaperone in Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 97–116. [Google Scholar] [CrossRef]
- Laurini, E.; Col, V.D.; Mamolo, M.G.; Zampieri, D.; Posocco, P.; Fermeglia, M.; Vio, L.; Pricl, S. Homology Model and Docking-Based Virtual Screening for Ligands of the Σ1 Receptor. ACS Med. Chem. Lett. 2011, 2, 834–839. [Google Scholar] [CrossRef]
- Laurini, E.; Marson, D.; Fermeglia, M.; Pricl, S. 3D Homology Model of Sigma1 Receptor. Handb. Exp. Pharmacol. 2017, 244, 27–50. [Google Scholar] [PubMed]
- Mavlyutov, T.A.; Yang, H.; Epstein, M.L.; Ruoho, A.E.; Yang, J.; Guo, L.-W. APEX2-Enhanced Electron Microscopy Distinguishes Sigma-1 Receptor Localization in the Nucleoplasmic Reticulum. Oncotarget 2017, 8, 51317. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Banerjee, D.; Ben-Baruch, A. Chemokines at the Crossroads of Tumor-Fibroblast Interactions That Promote Malignancy. J. Leukoc. Biol. 2011, 89, 31–39. [Google Scholar] [CrossRef]
- Yano, H.; Bonifazi, A.; Xu, M.; Guthrie, D.A.; Schneck, S.N.; Abramyan, A.M.; Fant, A.D.; Hong, W.C.; Newman, A.H.; Shi, L. Pharmacological Profiling of Sigma 1 Receptor Ligands by Novel Receptor Homomer Assays. Neuropharmacology 2018, 133, 264–275. [Google Scholar] [CrossRef]
- Maurice, T.; Goguadze, N. Sigma-1 (σ(1)) Receptor in Memory and Neurodegenerative Diseases. Handb. Exp. Pharmacol. 2017, 244, 81–108. [Google Scholar] [PubMed]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.; Kaushal, N.; Matsumoto, R.R. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. Adv. Exp. Med. Biol. 2017, 964, 133–152. [Google Scholar] [PubMed]
- Ortega-Roldan, J.L.; Ossa, F.; Schnell, J.R. Characterization of the Human Sigma-1 Receptor Chaperone Domain Structure and Binding Immunoglobulin Protein (BiP) Interactions. J. Biol. Chem. 2013, 288, 21448–21457. [Google Scholar] [CrossRef] [PubMed]
- Delprat, B.; Crouzier, L.; Su, T.-P.; Maurice, T. At the Crossing of ER Stress and MAMs: A Key Role of Sigma-1 Receptor? In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2020; Volume 1131, pp. 699–718. ISBN 978-3-030-12456-4. [Google Scholar]
- Mitsuda, T.; Omi, T.; Tanimukai, H.; Sakagami, Y.; Tagami, S.; Okochi, M.; Kudo, T.; Takeda, M. Sigma-1Rs Are Upregulated via PERK/EIF2α/ATF4 Pathway and Execute Protective Function in ER Stress. Biochem. Biophys. Res. Commun. 2011, 415, 519–525. [Google Scholar] [CrossRef]
- Hayashi, T. The Sigma-1 Receptor in Cellular Stress Signaling. Front. Neurosci. 2019, 13, 733. [Google Scholar] [CrossRef]
- Tsai, S.-Y.A.; Pokrass, M.J.; Klauer, N.R.; Nohara, H.; Su, T.-P. Sigma-1 Receptor Regulates Tau Phosphorylation and Axon Extension by Shaping P35 Turnover via Myristic Acid. Proc. Natl. Acad. Sci. USA 2015, 112, 6742–6747. [Google Scholar] [CrossRef]
- Miki, Y.; Mori, F.; Kon, T.; Tanji, K.; Toyoshima, Y.; Yoshida, M.; Sasaki, H.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Accumulation of the Sigma-1 Receptor Is Common to Neuronal Nuclear Inclusions in Various Neurodegenerative Diseases. Neuropathology 2014, 34, 148–158. [Google Scholar] [CrossRef]
- Vilner, B.J.; John, C.S.; Bowen, W.D. Sigma-1 and Sigma-2 Receptors Are Expressed in a Wide Variety of Human and Rodent Tumor Cell Lines. Cancer Res. 1995, 55, 408–413. [Google Scholar]
- Aydar, E.; Onganer, P.; Perrett, R.; Djamgoz, M.B.; Palmer, C.P. The Expression and Functional Characterization of Sigma (Sigma) 1 Receptors in Breast Cancer Cell Lines. Cancer Lett. 2006, 242, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Vilner, B.J.; de Costa, B.R.; Bowen, W.D. Cytotoxic Effects of Sigma Ligands: Sigma Receptor-Mediated Alterations in Cellular Morphology and Viability. J. Neurosci. 1995, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Spruce, B.A.; Campbell, L.A.; McTavish, N.; Cooper, M.A.; Appleyard, M.V.L.; O’Neill, M.; Howie, J.; Samson, J.; Watt, S.; Murray, K.; et al. Small Molecule Antagonists of the Sigma-1 Receptor Cause Selective Release of the Death Program in Tumor and Self-Reliant Cells and Inhibit Tumor Growth in Vitro and in Vivo. Cancer Res. 2004, 64, 4875–4886. [Google Scholar] [CrossRef] [PubMed]
- Berthois, Y.; Bourrié, B.; Galiègue, S.; Vidal, H.; Carayon, P.; Martin, P.M.; Casellas, P. SR31747A Is a Sigma Receptor Ligand Exhibiting Antitumoural Activity Both in Vitro and in Vivo. Br. J. Cancer 2003, 88, 438–446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, Q.-X.; Li, E.-M.; Zhang, Y.-F.; Liao, L.-D.; Xu, X.-E.; Wu, Z.-Y.; Shen, J.-H.; Xu, L.-Y. Overexpression of Sigma1 Receptor and Its Positive Associations with Pathologic TNM Classification in Esophageal Squamous Cell Carcinoma. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2012, 60, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, N.K.; Rybczynska, A.A.; Visser, A.K.D.; Marosi, K.; Nyakas, C.J.; Kwizera, C.; Sijbesma, J.W.A.; Elsinga, P.H.; Ishiwata, K.; Pruim, J.; et al. Small-Animal PET with a σ-Ligand, 11C-SA4503, Detects Spontaneous Pituitary Tumors in Aged Rats. J. Nucl. Med. 2013, 54, 1377–1383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Villemain, L.; Prigent, S.; Abou-Lovergne, A.; Pelletier, L.; Chiral, M.; Pontoglio, M.; Foufelle, F.; Caruso, S.; Pineau, R.; Rebouissou, S.; et al. Sigma 1 Receptor Is Overexpressed in Hepatocellular Adenoma: Involvement of ERα and HNF1α. Cancers 2020, 12, 2213. [Google Scholar] [CrossRef]
- Simony-Lafontaine, J.; Esslimani, M.; Bribes, E.; Gourgou, S.; Lequeux, N.; Lavail, R.; Grenier, J.; Kramar, A.; Casellas, P. Immunocytochemical Assessment of Sigma-1 Receptor and Human Sterol Isomerase in Breast Cancer and Their Relationship with a Series of Prognostic Factors. Br. J. Cancer 2000, 82, 1958–1966. [Google Scholar]
- Xu, Q.; Li, L.; Han, C.; Wei, L.; Kong, L.; Lin, F. Sigma-1 Receptor (Σ1R) Is Downregulated in Hepatic Malignant Tumors and Regulates HepG2 Cell Proliferation, Migration and Apoptosis. Oncol. Rep. 2018, 39, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- van Waarde, A.; Rybczynska, A.A.; Ramakrishnan, N.K.; Ishiwata, K.; Elsinga, P.H.; Dierckx, R.A.J.O. Potential Applications for Sigma Receptor Ligands in Cancer Diagnosis and Therapy. Biochim. Biophys. Acta 2015, 1848, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.J.; Maher, C.M. Sigma1 Pharmacology in the Context of Cancer. Handb. Exp. Pharmacol. 2017, 244, 237–308. [Google Scholar] [PubMed]
- Kim, F.J.; Schrock, J.M.; Spino, C.M.; Marino, J.C.; Pasternak, G.W. Inhibition of Tumor Cell Growth by Sigma1 Ligand Mediated Translational Repression. Biochem. Biophys. Res. Commun. 2012, 426, 177–182. [Google Scholar] [CrossRef]
- Achison, M.; Boylan, M.T.; Hupp, T.R.; Spruce, B.A. HIF-1α Contributes to Tumour-Selective Killing by the Sigma Receptor Antagonist Rimcazole. Oncogene 2007, 26, 1137–1146. [Google Scholar] [CrossRef]
- Schrock, J.M.; Spino, C.M.; Longen, C.G.; Stabler, S.M.; Marino, J.C.; Pasternak, G.W.; Kim, F.J. Sequential Cytoprotective Responses to Sigma1 Ligand-Induced Endoplasmic Reticulum Stress. Mol. Pharmacol. 2013, 84, 751–762. [Google Scholar] [CrossRef]
- Do, W.; Herrera, C.; Mighty, J.; Shumskaya, M.; Redenti, S.M.; Sauane, M. Sigma 1 Receptor Plays a Prominent Role in IL-24-Induced Cancer-Specific Apoptosis. Biochem. Biophys. Res. Commun. 2013, 439, 215–220. [Google Scholar] [CrossRef]
- Marriott, K.-S.C.; Prasad, M.; Thapliyal, V.; Bose, H.S. σ-1 Receptor at the Mitochondrial-Associated Endoplasmic Reticulum Membrane Is Responsible for Mitochondrial Metabolic Regulation. J. Pharmacol. Exp. Ther. 2012, 343, 578–586. [Google Scholar] [CrossRef]
- Hayashi, T.; Hayashi, E.; Fujimoto, M.; Sprong, H.; Su, T.-P. The Lifetime of UDP-Galactose:Ceramide Galactosyltransferase is Controlled by a Distinct Endoplasmic Reticulum-Associated Degradation (ERAD) Regulated by Sigma-1 Receptor Chaperones. J. Biol. Chem. 2012, 287, 43156–43169. [Google Scholar] [CrossRef]
- Litan, A.; Langhans, S.A. Cancer as a Channelopathy: Ion Channels and Pumps in Tumor Development and Progression. Front. Cell. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef]
- Crociani, O.; Zanieri, F.; Pillozzi, S.; Lastraioli, E.; Stefanini, M.; Fiore, A.; Fortunato, A.; D’Amico, M.; Masselli, M.; De Lorenzo, E.; et al. HERG1 Channels Modulate Integrin Signaling to Trigger Angiogenesis and Tumor Progression in Colorectal Cancer. Sci. Rep. 2013, 3, 3308. [Google Scholar] [CrossRef]
- Masi, A.; Becchetti, A.; Restano-Cassulini, R.; Polvani, S.; Hofmann, G.; Buccoliero, A.M.; Paglierani, M.; Pollo, B.; Taddei, G.L.; Gallina, P.; et al. HERG1 Channels Are Overexpressed in Glioblastoma Multiforme and Modulate VEGF Secretion in Glioblastoma Cell Lines. Br. J. Cancer 2005, 93, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Brizzi, M.F.; Bernabei, P.A.; Bartolozzi, B.; Caporale, R.; Basile, V.; Boddi, V.; Pegoraro, L.; Becchetti, A.; Arcangeli, A. VEGFR-1 (FLT-1), Beta1 Integrin, and HERG K+ Channel for a Macromolecular Signaling Complex in Acute Myeloid Leukemia: Role in Cell Migration and Clinical Outcome. Blood 2007, 110, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Masselli, M.; De Lorenzo, E.; Accordi, B.; Cilia, E.; Crociani, O.; Amedei, A.; Veltroni, M.; D’Amico, M.; Basso, G.; et al. Chemotherapy Resistance in Acute Lymphoblastic Leukemia Requires HERG1 Channels and is Overcome by HERG1 Blockers. Blood 2011, 117, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Crottès, D.; Rapetti-Mauss, R.; Alcaraz-Perez, F.; Tichet, M.; Gariano, G.; Martial, S.; Guizouarn, H.; Pellissier, B.; Loubat, A.; Popa, A.; et al. SIGMAR1 Regulates Membrane Electrical Activity in Response to Extracellular Matrix Stimulation to Drive Cancer Cell Invasiveness. Cancer Res. 2016, 76, 607–618. [Google Scholar] [CrossRef]
- Balasuriya, D.; Stewart, A.P.; Crottès, D.; Borgese, F.; Soriani, O.; Edwardson, J.M. The Sigma-1 Receptor Binds to the Nav1.5 Voltage-Gated Na+ Channel with 4-Fold Symmetry. J. Biol. Chem. 2012, 287, 37021–37029. [Google Scholar] [CrossRef]
- Brisson, L.; Gillet, L.; Calaghan, S.; Besson, P.; Le Guennec, J.-Y.; Roger, S.; Gore, J. Na(V)1.5 Enhances Breast Cancer Cell Invasiveness by Increasing NHE1-Dependent H(+) Efflux in Caveolae. Oncogene 2011, 30, 2070–2076. [Google Scholar] [CrossRef]
- Renaudo, A.; L’Hoste, S.; Guizouarn, H.; Borgèse, F.; Soriani, O. Cancer Cell Cycle Modulated by a Functional Coupling between Sigma-1 Receptors and Cl- Channels. J. Biol. Chem. 2007, 282, 2259–2267. [Google Scholar] [CrossRef]
- Ela, C.; Barg, J.; Vogel, Z.; Hasin, Y.; Eilam, Y. Sigma Receptor Ligands Modulate Contractility, Ca++ Influx and Beating Rate in Cultured Cardiac Myocytes. J. Pharmacol. Exp. Ther. 1994, 269, 1300–1309. [Google Scholar]
- Hayashi, T.; Kagaya, A.; Takebayashi, M.; Shimizu, M.; Uchitomi, Y.; Motohashi, N.; Yamawaki, S. Modulation by Sigma Ligands of Intracellular Free Ca++ Mobilization by N-Methyl-D-Aspartate in Primary Culture of Rat Frontal Cortical Neurons. J. Pharmacol. Exp. Ther. 1995, 275, 207–214. [Google Scholar]
- Klette, K.L.; Lin, Y.; Clapp, L.E.; DeCoster, M.A.; Moreton, J.E.; Tortella, F.C. Neuroprotective Sigma Ligands Attenuate NMDA and Trans-ACPD-Induced Calcium Signaling in Rat Primary Neurons. Brain Res. 1997, 756, 231–240. [Google Scholar] [CrossRef]
- Monnet, F.P.; Morin-Surun, M.P.; Leger, J.; Combettes, L. Protein Kinase C-Dependent Potentiation of Intracellular Calcium Influx by Sigma1 Receptor Agonists in Rat Hippocampal Neurons. J. Pharmacol. Exp. Ther. 2003, 307, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Regulating Ankyrin Dynamics: Roles of Sigma-1 Receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Bowen, W.D. Role of Sigma-1 Receptor C-Terminal Segment in Inositol 1,4,5-Trisphosphate Receptor Activation: Constitutive Enhancement of Calcium Signaling in MCF-7 Tumor Cells. J. Biol. Chem. 2008, 283, 28198–28215. [Google Scholar] [CrossRef] [PubMed]
- Abou-Lovergne, A.; Collado-Hilly, M.; Monnet, F.P.; Koukoui, O.; Prigent, S.; Coquil, J.F.; Dupont, G.; Combettes, L. Investigation of the Role of Sigma1-Receptors in Inositol 1,4,5-Trisphosphate Dependent Calcium Signaling in Hepatocytes. Cell Calcium 2011, 50, 62–72. [Google Scholar] [CrossRef]
- Srivats, S.; Balasuriya, D.; Pasche, M.; Vistal, G.; Edwardson, J.M.; Taylor, C.W.; Murrell-Lagnado, R.D. Sigma1 Receptors Inhibit Store-Operated Ca2+ Entry by Attenuating Coupling of STIM1 to Orai1. J. Cell Biol. 2016, 213, 65–79. [Google Scholar] [CrossRef]
- Gueguinou, M.; Crottès, D.; Chantôme, A.; Rapetti-Mauss, R.; Potier-Cartereau, M.; Clarysse, L.; Girault, A.; Fourbon, Y.; Jézéquel, P.; Guérin-Charbonnel, C.; et al. The SigmaR1 Chaperone Drives Breast and Colorectal Cancer Cell Migration by Tuning SK3-Dependent Ca(2+) Homeostasis. Oncogene 2017, 36, 3640–3647. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pontisso, I.; Combettes, L. Role of Sigma-1 Receptor in Calcium Modulation: Possible Involvement in Cancer. Genes 2021, 12, 139. https://doi.org/10.3390/genes12020139
Pontisso I, Combettes L. Role of Sigma-1 Receptor in Calcium Modulation: Possible Involvement in Cancer. Genes. 2021; 12(2):139. https://doi.org/10.3390/genes12020139
Chicago/Turabian StylePontisso, Ilaria, and Laurent Combettes. 2021. "Role of Sigma-1 Receptor in Calcium Modulation: Possible Involvement in Cancer" Genes 12, no. 2: 139. https://doi.org/10.3390/genes12020139
APA StylePontisso, I., & Combettes, L. (2021). Role of Sigma-1 Receptor in Calcium Modulation: Possible Involvement in Cancer. Genes, 12(2), 139. https://doi.org/10.3390/genes12020139