
Kinase Inhibitors of DNA-PK, ATM and ATR in Combination with Ionizing Radiation Can Increase Tumor Cell Death in HNSCC Cells While Sparing Normal Tissue Cells


 ,
,  and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Culture and Inhibitors
2.2. Flow Cytometry Analysis of Apoptosis and Necrosis
2.3. Flow Cytometry Analysis of Cell Cycle
2.4. Wound Healing Assay
2.5. Colony Forming Assay
2.6. Statistical Analysis
2.7. Ethics Approval and Consent to Participate
3. Results
3.1. Concomitant KI and IR Treatment Leads to Increase of Cell Death
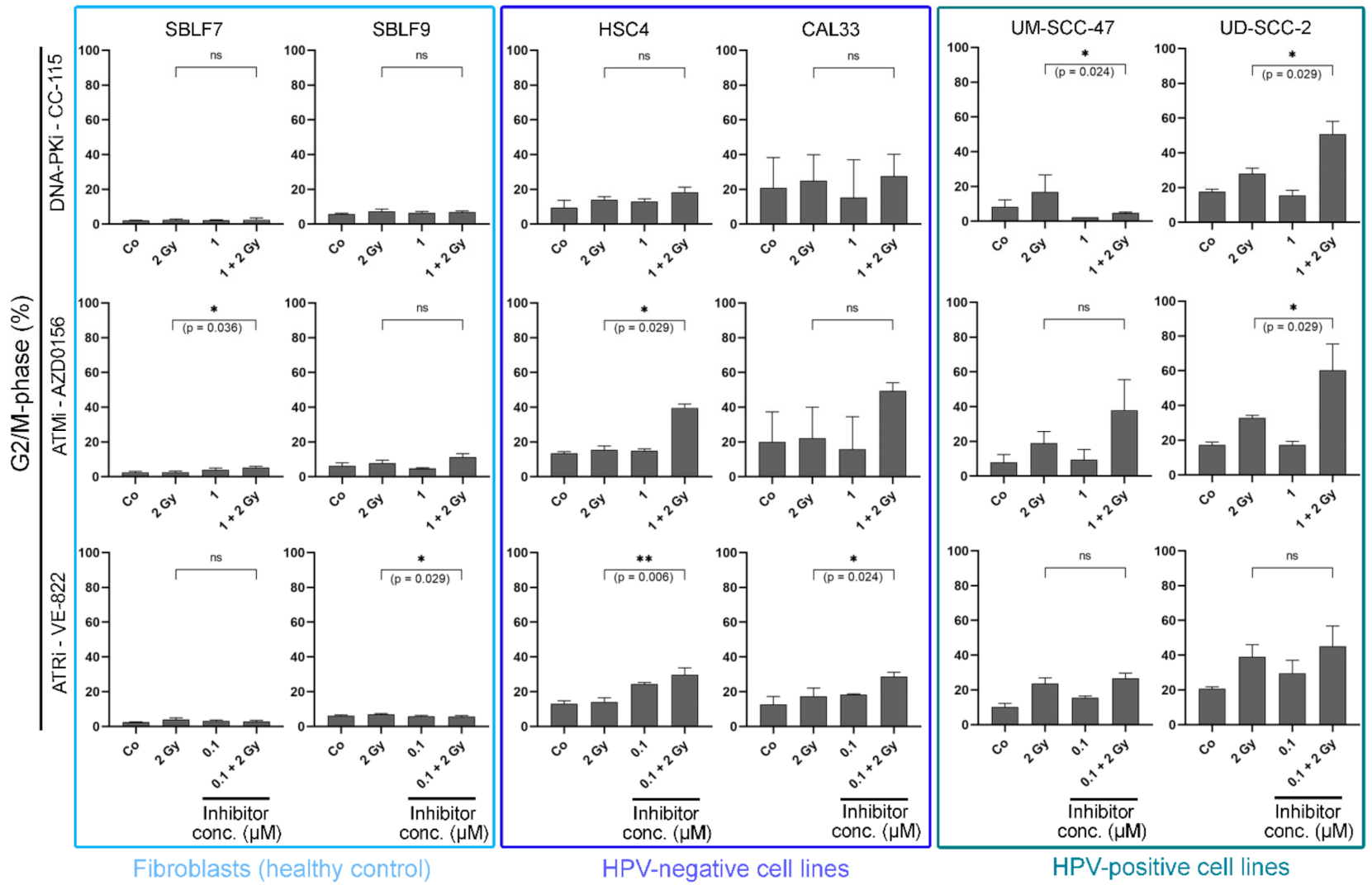
3.2. KI Mainly Arrests Cancer Cells in G2/M
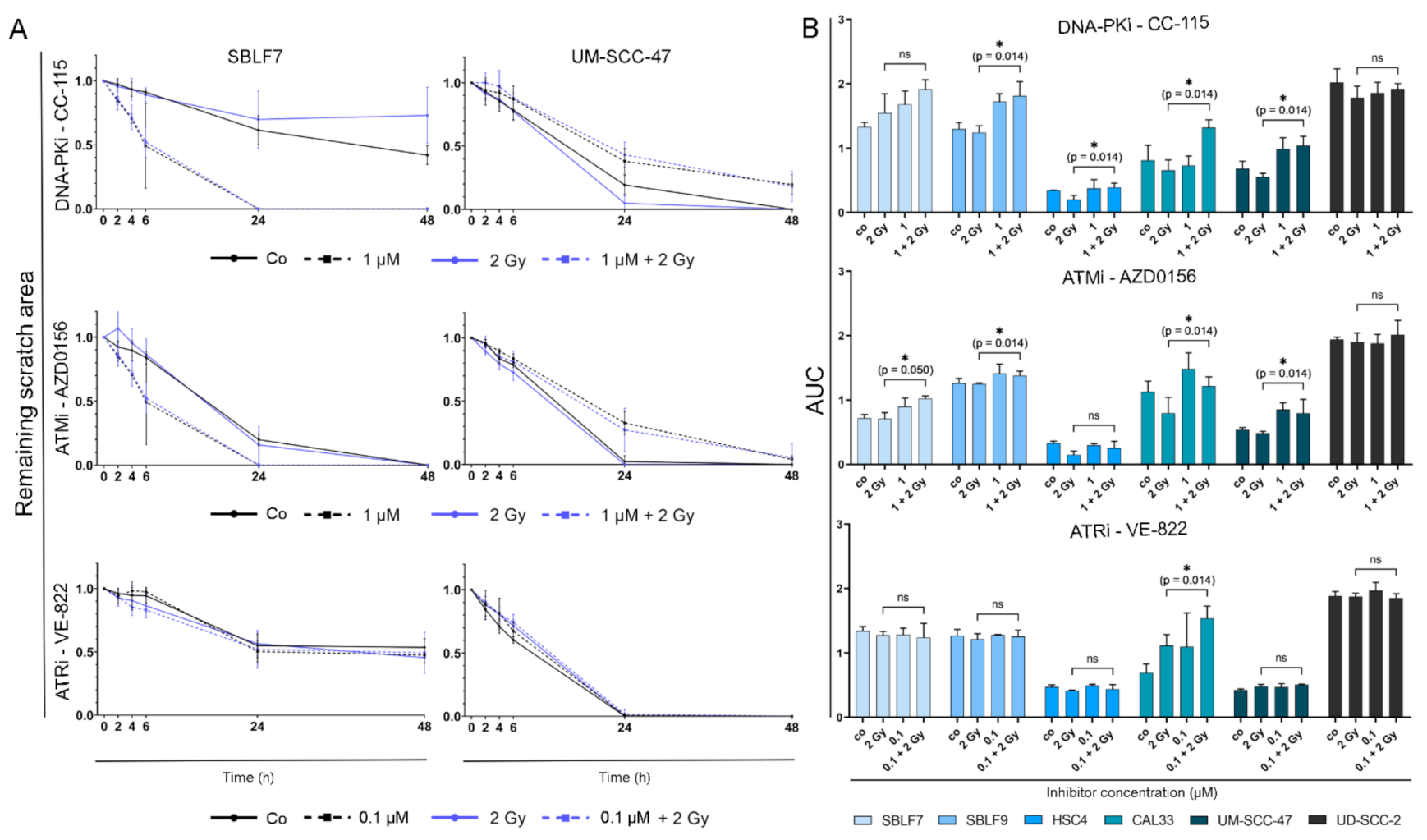
3.3. Combined Treatment Synergistically Reduces HNSCC Cell Survival
3.4. Migration Behavior of HNSCC Cell Lines Is Strongly Influenced by Inter-Individual Differences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Brandsma, I.; Fleuren, E.D.; Williamson, C.T.; Lord, C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef]
- Glorieux, M.; Dok, R.; Nuyts, S. Novel DNA targeted therapies for head and neck cancers: Clinical potential and biomarkers. Oncotarget 2017, 8, 81662–81678. [Google Scholar] [CrossRef] [PubMed]
- Saberi, A.; Hochegger, H.; Szuts, D.; Lan, L.; Yasui, A.; Sale, J.E.; Taniguchi, Y.; Murakawa, Y.; Zeng, W.; Yokomori, K.; et al. RAD18 and Poly(ADP-Ribose) Polymerase Independently Suppress the Access of Nonhomologous End Joining to Double-Strand Breaks and Facilitate Homologous Recombination-Mediated Repair. Mol. Cell. Biol. 2007, 27, 2562–2571. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Raghavan, S.C. DNA Double-Strand Break Repair Inhibitors as Cancer Therapeutics. Chem. Biol. 2015, 22, 17–29. [Google Scholar] [CrossRef]
- Basourakos, S.P.; Li, L.; Aparicio, A.M.; Corn, P.G.; Kim, J.; Thompson, T.C. Combination Platinum-based and DNA Damage Response-targeting Cancer Therapy: Evolution and Future Directions. Curr. Med. Chem. 2017, 24, 1586–1606. [Google Scholar] [CrossRef]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-in-Human Phase I Study of a Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) in Advanced Malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 1–27. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 1–22. [Google Scholar] [CrossRef]
- Hecht, M.; Meier, F.; Zimmer, L.; Polat, B.; Loquai, C.; Weishaupt, C.; Forschner, A.; Gutzmer, R.; Utikal, J.; Goldinger, S.M.; et al. Clinical outcome of concomitant vs interrupted BRAF inhibitor therapy during radiotherapy in melanoma patients. Br. J. Cancer 2018, 118, 785–792. [Google Scholar] [CrossRef]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Castilho, R.M.; Squarize, C.H.; Almeida, L.O. Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. Int. J. Mol. Sci. 2017, 18, 1506. [Google Scholar] [CrossRef]
- Fabbrizi, M.R.; Parsons, J.L. Radiotherapy and the cellular DNA damage response: Current and future perspectives on head and neck cancer treatment. Cancer Drug Resist. 2020, 3, 775–790. [Google Scholar] [CrossRef]
- Economopoulou, P.; Perisanidis, C.; Giotakis, E.I.; Psyrri, A. The emerging role of immunotherapy in head and neck squamous cell carcinoma (HNSCC): Anti-tumor immunity and clinical applications. Ann. Transl. Med. 2016, 4, 173. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Dodhia, S.; Su, G. Dysregulations in the PI3K pathway and targeted therapies for head and neck squamous cell carcinoma. Oncotarget 2017, 8, 22203–22217. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Shen, J.; Weems, A.; Lu, S.-L. Role of Phosphatidylinositol-3-Kinase Pathway in Head and Neck Squamous Cell Carcinoma. J. Oncol. 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Cortez, D. Common mechanisms of PIKK regulation. DNA Repair. 2009, 8, 1004–1008. [Google Scholar] [CrossRef]
- Bürkel, F.; Jost, T.; Hecht, M.; Heinzerling, L.; Fietkau, R.; Distel, L. Dual mTOR/DNA-PK Inhibitor CC-115 Induces Cell Death in Melanoma Cells and Has Radiosensitizing Potential. Int. J. Mol. Sci. 2020, 21, 9321. [Google Scholar] [CrossRef] [PubMed]
- Weigert, V.; Jost, T.; Hecht, M.; Knippertz, I.; Heinzerling, L.; Fietkau, R.; Distel, L.V. PARP inhibitors combined with ionizing radiation induce different effects in melanoma cells and healthy fibroblasts. BMC Cancer 2020, 20, 775. [Google Scholar] [CrossRef]
- Kang, M.A.; Kim, W.; Jo, H.-R.; Shin, Y.-J.; Kim, M.-H.; Jeong, J.-H. Anticancer and radiosensitizing effects of the cyclin-dependent kinase inhibitors, AT7519 and SNS-032, on cervical cancer. Int. J. Oncol. 2018, 53, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Vávrová, J.; Zárybnická, L.; Jošt, P.; Tichý, A.; Řezáčová, M.; Šinkorová, Z.; Pejchal, J. Comparison of the Radiosensitizing Effect of ATR, ATM and DNA-PK Kinase Inhibitors on Cervical Carcinoma Cells. Folia Biol. 2016, 62, 167–174. [Google Scholar]
- Merten, R.; Hecht, M.; Haderlein, M.; Distel, L.; Fietkau, R.; Heinzerling, L.; Semrau, S. Increased skin and mucosal toxicity in the combination of vemurafenib with radiation therapy. Strahlenther. Onkol. 2014, 190, 1169–1172. [Google Scholar] [CrossRef]
- Satzger, I.; Degen, A.; Asper, H.; Kapp, A.; Hauschild, A.; Gutzmer, R. Serious Skin Toxicity With the Combination of BRAF Inhibitors and Radiotherapy. J. Clin. Oncol. 2013, 31, e220–e222. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Thomsen, A.; Follo, M.; Zamboglou, C.; Bronsert, P.; Mostafa, H.; Amen, A.; Mekawy, M.; Grosu, A.L.; Brunner, T.B. FAK inhibition radiosensitizes pancreatic ductal adenocarcinoma cells in vitro. Strahlenther. Onkol. 2021, 197, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jin, Y.; Ying, H.; Zhang, P.; Chen, M.; Hu, X. Synergistic effect of PAF inhibition and X-ray irradiation in non-small cell lung cancer cells. Strahlenther. Onkol. 2021, 197, 343–352. [Google Scholar] [CrossRef]
- Hecht, M.; Harrer, T.; Körber, V.; Sarpong, E.O.; Moser, F.; Fiebig, N.; Schwegler, M.; Stürzl, M.; Fietkau, R.; Distel, L. Cytotoxic effect of Efavirenz in BxPC-3 pancreatic cancer cells is based on oxidative stress and is synergistic with ionizing radiation. Oncol. Lett. 2017, 15, 1728–1736. [Google Scholar] [CrossRef]
- Sinclair, W.K. Cyclic X-ray Responses in Mammalian Cellsin Vitro. Radiat. Res. 1968, 33, 620–643. [Google Scholar] [CrossRef] [PubMed]
- Gryc, T.; Putz, F.; Goerig, N.; Ziegler, S.; Fietkau, R.; Distel, L.V.; Schuster, B. Idelalisib may have the potential to increase radiotherapy side effects. Radiat. Oncol. 2017, 12, 109. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Hendrickson, A.E.W.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Fietkau, R.; Hecht, M.; Hofner, B.; Lubgan, D.; Iro, H.; Gefeller, O.; Rödel, C.; Hautmann, M.G.; Kölbl, O.; Salay, A.; et al. Randomized phase-III-trial of concurrent chemoradiation for locally advanced head and neck cancer comparing dose reduced radiotherapy with paclitaxel/cisplatin to standard radiotherapy with fluorouracil/cisplatin: The PacCis-trial. Radiother. Oncol. 2020, 144, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, F.; Minafra, L.; Cammarata, F.P.; Savoca, G.; Calvaruso, M.; Vicario, N.; Maccari, L.; Pérès, E.A.; Özçelik, H.; Bernaudin, M.; et al. SRC Tyrosine Kinase Inhibitor and X-rays Combined Effect on Glioblastoma Cell Lines. Int. J. Mol. Sci. 2020, 21, 3917. [Google Scholar] [CrossRef]
- Katz, D.; Ito, E.; Lau, K.S.; Mocanu, J.D.; Bastianutto, C.; Schimmer, A.D.; Liu, F.-F. Increased efficiency for performing colony formation assays in 96-well plates: Novel applications to combination therapies and high-throughput screening. Biotechniques 2008, 44, ix–xiv. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Regulski, M.J. Cellular senescence: What, why, and how. Wounds 2017, 29, 74–168. [Google Scholar] [PubMed]
- Dobler, C.; Jost, T.; Hecht, M.; Fietkau, R.; Distel, L. Senescence Induction by Combined Ionizing Radiation and DNA Damage Response Inhibitors in Head and Neck Squamous Cell Carcinoma Cells. Cells 2020, 9, 2012. [Google Scholar] [CrossRef]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef]
- Yan, Y.; Black, C.P.; Cowan, K.H. Irradiation-induced G2/M checkpoint response requires ERK1/2 activation. Oncogene 2007, 26, 4689–4698. [Google Scholar] [CrossRef]
- Mc Strasser-Wozak, E.; Hartmann, B.L.; Geley, S.; Sgonc, R.; Böck, G.; Dos Santos, A.J.O.; Hattmannstorfer, R.; Wolf, H.; Pavelka, M.; Kofler, R. Irradiation induces G2/M cell cycle arrest and apoptosis in p53-deficient lymphoblastic leukemia cells without affecting Bcl-2 and Bax expression. Cell Death Differ. 1998, 5, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Willmore, E.; De Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.A.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef]
- Zhao, Y.; Thomas, H.D.; Batey, M.A.; Cowell, I.; Richardson, C.J.; Griffin, R.J.; Calvert, A.H.; Newell, D.R.; Smith, G.C.; Curtin, N.J. Preclinical Evaluation of a Potent Novel DNA-Dependent Protein Kinase Inhibitor NU7441. Cancer Res. 2006, 66, 5354–5362. [Google Scholar] [CrossRef]
- Sturgeon, C.M.; Knight, Z.; Shokat, K.M.; Roberge, M. Effect of combined DNA repair inhibition and G2 checkpoint inhibition on cell cycle progression after DNA damage. Mol. Cancer Ther. 2006, 5, 885–892. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seifrtová, M.; Havelek, R.; Chmelařová, M.; Cmielová, J.; Muthná, D.; Stoklasová, A.; Zemankova, S.; Rezacova, M. The effect of ATM and ERK1/2 inhibition on mitoxantrone-induced cell death of leukaemic cells. Folia Biol. 2011, 57, 74–81. [Google Scholar]
- Zhu, H.; Miao, Z.-H.; Huang, M.; Feng, J.-M.; Zhang, Z.-X.; Lu, J.; Cai, Y.-J.; Tong, L.-J.; Xu, Y.-F.; Qian, X.-H.; et al. Naphthalimides Induce G2 Arrest through the ATM-Activated Chk2-Executed Pathway in HCT116 Cells. Neoplasia 2009, 11, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Arlander, S.J.H.; Greene, B.T.; Innes, C.L.; Paules, R.S. DNA Protein Kinase–Dependent G2 Checkpoint Revealed following Knockdown of Ataxia-Telangiectasia Mutated in Human Mammary Epithelial Cells. Cancer Res. 2008, 68, 89–97. [Google Scholar] [CrossRef]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, F.; Mathew, L.M.; Schwartz, R.A. Radiation dermatitis: An overview. Int. J. Dermatol. 2017, 56, 909–914. [Google Scholar] [CrossRef]
- Maria, O.M.; Eliopoulos, N.; Muanza, T. Radiation-Induced Oral Mucositis. Front. Oncol. 2017, 7, 89. [Google Scholar] [CrossRef]
- Hecht, M.; Hahn, D.; Wolber, P.; Hautmann, M.G.; Reichert, D.; Weniger, S.; Belka, C.; Bergmann, T.; Göhler, T.; Welslau, M.; et al. Treatment response lowers tumor symptom burden in recurrent and/or metastatic head and neck cancer. BMC Cancer 2020, 20, 933. [Google Scholar] [CrossRef]
- Van der Kamp, M.F.; Muntinghe, F.O.W.; Iepsma, R.S.; Plaat, B.E.C.; van der Laan, B.F.A.M.; Algassab, A.; Steenbakkers, R.J.H.M.; Witjes, M.; van Dijk, B.A.C.; de Bock, G.H.; et al. Predictors for distant metastasis in head and neck cancer, with emphasis on age. Eur. Arch. Otorhinolaryngol. 2021, 278, 181–190. [Google Scholar] [CrossRef]
- Bhave, S.L.; Teknos, T.N.; Pan, Q. Molecular Parameters of Head and Neck Cancer Metastasis. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 143–153. [Google Scholar] [CrossRef]
- Pisani, E.B.P.; Airoldi, M.; Allais, A.; Valletti, P.A.; Battista, M.; Benazzo, M.; Briatore, R.; Cacciola, S.; Cocuzza, S.; Colombo, A.; et al. Metastatic disease in head & neck oncology. Acta Otorhinolaryngol. Ital. 2020, 40 (Suppl. S1), S1–S86. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Horwitz, A.F. Cell Migration: A Physically Integrated Molecular Process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Pijuan, J.; Barceló, C.; Moreno, D.; Maiques, O.; Sisó, P.; Marti, R.M.; Macià, A.; Panosa, A. In vitro Cell Migration, Invasion, and Adhesion Assays: From Cell Imaging to Data Analysis. Front. Cell Dev. Biol. 2019, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Jandial, R. (Ed.) Metastatic Cancer: Clinical and Biological Perspectives; Molecular Biology Intelligence Unit; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Kidiyoor, G.R.; Li, Q.; Bastianello, G.; Bruhn, C.; Giovannetti, I.; Mohamood, A.; Beznoussenko, G.V.; Mironov, A.; Raab, M.; Piel, M.; et al. ATR is essential for preservation of cell mechanics and nuclear integrity during interstitial migration. Nat. Commun. 2020, 11, 4828. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, A.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Broustas, C.; Lieberman, H.B. DNA Damage Response Genes and the Development of Cancer Metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef]
- Yang, H.; Yao, F.; Marti, T.M.; Schmid, R.A.; Peng, R.-W. Beyond DNA Repair: DNA-PKcs in Tumor Metastasis, Metabolism and Immunity. Cancers 2020, 12, 3389. [Google Scholar] [CrossRef] [PubMed]
- Medová, M.; Medo, M.; Hovhannisyan, L.; Muñoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in human malignant disorders: Mechanisms and implications for pharmacological interventions. Pharmacol. Ther. 2020, 215, 107617. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.-C.; Fang, P.-T.; Lee, Y.-C.; Wang, Y.-Y.; Su, Y.-H.; Hu, S.C.-S.; Chen, Y.-K.; Tsui, Y.-T.; Kao, Y.-H.; Huang, M.-Y.; et al. DNA Repair Protein Rad51 Induces Tumor Growth and Metastasis in Esophageal Squamous Cell Carcinoma via a p38/Akt-Dependent Pathway. Ann. Surg. Oncol. 2020, 27, 2090–2101. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Pan, L.; Kang, N.; Shen, X. PP4R1 interacts with HMGA2 to promote non-small-cell lung cancer migration and metastasis via activating MAPK/ERK-induced epithelial-mesenchymal transition. Mol. Carcinog. 2020, 59, 467–477. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors | ||||||
|---|---|---|---|---|---|---|
| CC-115 | AZD0156 | VE-822 | ||||
| Cell lines | 0 Gy | 2 Gy | 0 Gy | 2 Gy | 0 Gy | 2 Gy |
| SBLF7 | 1.0 * c + 6.4 | 0.8 * c + 8.7 | 0.7 * c + 12.1 | 0.2 * c + 12.2 | 2.0 * c + 9.2 | 3.9 * c + 9.7 |
| SBLF9 | −0.7 * c + 16.2 | −2.4 * c + 19.7 | 2.0 * c + 21.3 | 2.9 * c + 19.5 | 9.7 * c + 13.9 | 4.1 * c + 16.1 |
| HSC4 | 3.9 * c + 9.9 | 4.4 * c + 12.8 | 8.1 * c + 9.0 | 12.8 * c + 15.9 | 98.8 * c + 15.0 | 76.7 * c + 21.7 |
| CAL33 | 11.7 * c + 12.3 | 10.3 * c + 21.3 | 12.9 * c + 14.4 | 13.7 * c + 28.3 | 65.3 * c + 11.0 | 74.7 * c + 22.2 |
| UM-SCC-47 | 14.3 * c + 9.3 | 11.2 * c + 15.8 | 8.8 * c + 9.1 | 12.1 * c + 20.6 | 93.9 * c + 11.8 | 74.9 * c + 28.3 |
| UD-SCC-2 | 16.7 * c + 17.9 | 12.2 * c + 29.7 | 3.0 * c + 23.2 | 0.4 * c + 39.8 | 35.7 * c + 43.0 | 43.1 * c + 45.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faulhaber, E.-M.; Jost, T.; Symank, J.; Scheper, J.; Bürkel, F.; Fietkau, R.; Hecht, M.; Distel, L.V. Kinase Inhibitors of DNA-PK, ATM and ATR in Combination with Ionizing Radiation Can Increase Tumor Cell Death in HNSCC Cells While Sparing Normal Tissue Cells. Genes 2021, 12, 925. https://doi.org/10.3390/genes12060925
Faulhaber E-M, Jost T, Symank J, Scheper J, Bürkel F, Fietkau R, Hecht M, Distel LV. Kinase Inhibitors of DNA-PK, ATM and ATR in Combination with Ionizing Radiation Can Increase Tumor Cell Death in HNSCC Cells While Sparing Normal Tissue Cells. Genes. 2021; 12(6):925. https://doi.org/10.3390/genes12060925
Chicago/Turabian StyleFaulhaber, Eva-Maria, Tina Jost, Julia Symank, Julian Scheper, Felix Bürkel, Rainer Fietkau, Markus Hecht, and Luitpold V. Distel. 2021. "Kinase Inhibitors of DNA-PK, ATM and ATR in Combination with Ionizing Radiation Can Increase Tumor Cell Death in HNSCC Cells While Sparing Normal Tissue Cells" Genes 12, no. 6: 925. https://doi.org/10.3390/genes12060925
APA StyleFaulhaber, E.-M., Jost, T., Symank, J., Scheper, J., Bürkel, F., Fietkau, R., Hecht, M., & Distel, L. V. (2021). Kinase Inhibitors of DNA-PK, ATM and ATR in Combination with Ionizing Radiation Can Increase Tumor Cell Death in HNSCC Cells While Sparing Normal Tissue Cells. Genes, 12(6), 925. https://doi.org/10.3390/genes12060925