
Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge

 , ,
, ,
Abstract
1. Introduction
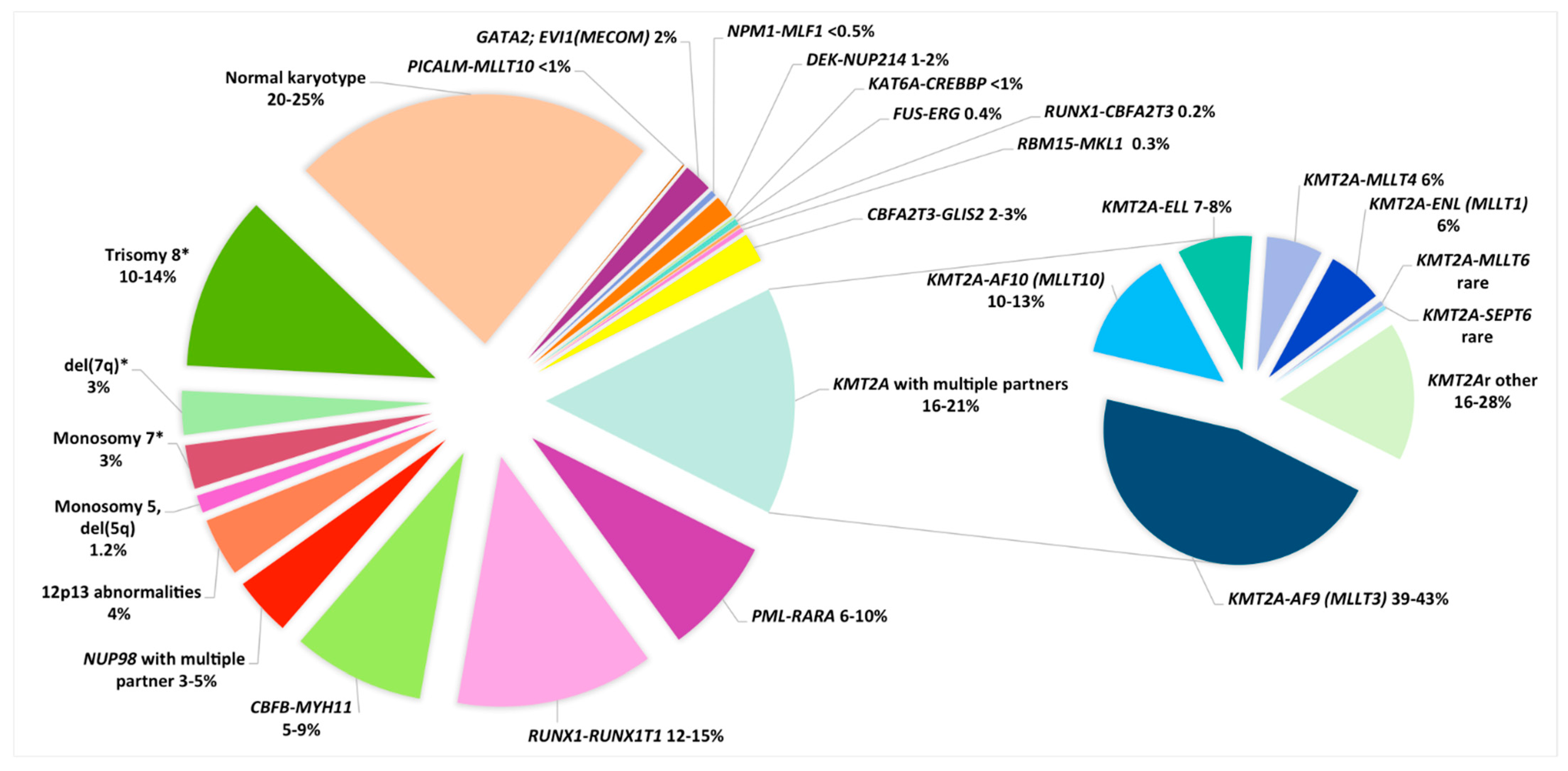
2. Cytogenetic Subgroups
2.1. Balanced Cytogenetic Abnormalities
2.1.1. Acute Promyelocytic Leukemia (APL), M3-M3v/t(15;17)(q24;q21), Now Referred to as APL with PML-RARA
2.1.2. Core Binding Factor AML
2.1.3. KMT2A/11q23 Rearrangements
2.1.4. 11p15/NUP98 Rearrangements
2.1.5. 12p Abnormalities Including the Rare t(7;12)(q36;p13)/ETV6;MNX1
2.2. Rare Balanced Rearrangements
2.2.1. Inversion (3;3)(q21q26.2)/t(3;3)(q21;q26.2)/GATA2;MECOM (EVI1)
2.2.2. Translocation (6;9)(p22;q34)/DEK-NUP214
2.2.3. Translocation t(3;5)(q25;q35)/NPM1-MLF1
2.2.4. Translocation t(8;16)(p11;p13)/KAT6A-CREBBP
2.2.5. t(16;21)(p11;q22)/FUS-ERG
2.2.6. t(16;21)(q24;q22)/RUNX1-CBFA2T3
2.2.7. Translocation (1;22)(p13;q13)/RBM15-MLK1
2.2.8. The Cryptic Inversion, inv(16)(p13.3q24.3)/CBFA2T3-GLIS2
2.3. Unbalanced Cytogenetic Abnormalities
2.3.1. Partial or Total Loss of Chromosomes
Monosomy 7 and del(7q)
Monosomy 5 and del(5q) (-5/5q-)
2.3.2. Gains of Chromosomes
Trisomy 8
2.3.3. Complex, Hyperdiploid and Monosomal Karyotypes
2.4. Normal Karyotypes

{kind=link}
{kind=link}
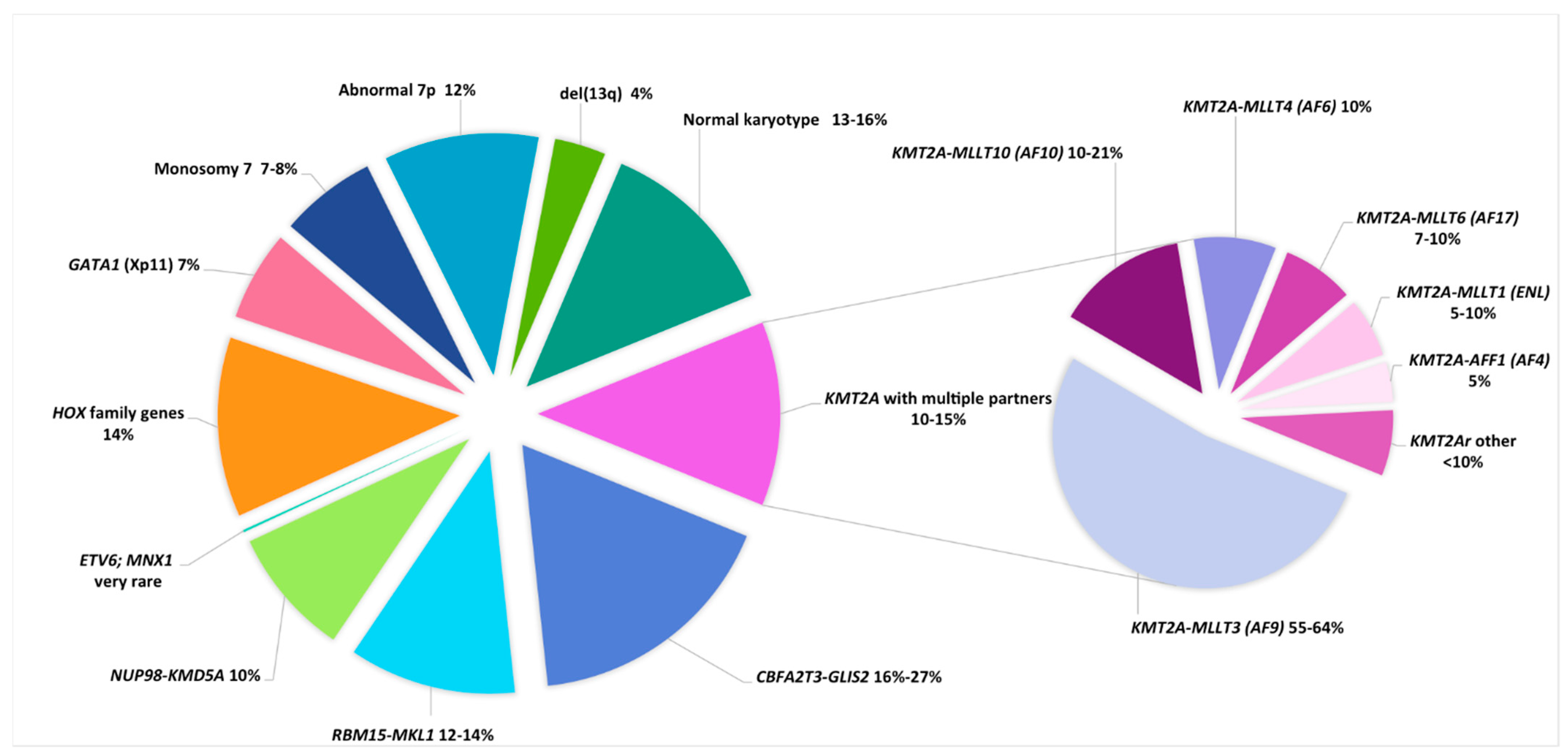
| Cytogenetic Subgroups | Fusion Gene or Genes Involved | Frequency in Non-DS AMKL | Median Age, (Range), years | Special Features | Secondary CA | Secondary Molecular Abnormalities | Prognosis | References |
|---|---|---|---|---|---|---|---|---|
| DS AMKL | ||||||||
| Trisomy 21c | GATA1 (Xp11) truncating mutation | NA | 1.7 (0.4–3.8) | 85–97% of DS-AML were M7 TAM in 25% of DS pts that can evolve towards M7 in 10% of cases | tri 8, gain of a third chr 21, gain of 1q | Mutations in cohesin complex genes (STAG2, RAD21, …), MPL, RAS, JAK2, JAK3 | Good (impaired by trisomy 8?) | [9,141,142,143,144] |
| Non-DS AMKL | 1.6 (0.1–17) | Mainly infants Hepatosplenomegaly Myelofibrosis that can impair sampling for diagnosis | [48,62,63,64] | |||||
| inv(16)(p13.3q24.3) * | CBFA2T3-GLIS2 | 20% (16–27%) | 1.5 (0.5–4) | Infants, extramedullary disease, CD56++ | tri 21 tri 3 | Low frequency of mutations | Very Poor | [48,63,64,65] |
| t(1;22)(p13;q13) | RBM15-MKL1 | 12–14% | 0.7 (0.1–2.7) | Only M7 Hepatosplenomegaly, Fibrosis | Mainly no ACA HD karyotypes, tri 1q (unbalanced t(1;22) in 26% of cases) | Low frequency of mutations | Intermediate | [48,60,61,62,63,64] |
| 11q23.3/KMT2r | KMT2A with multiple partners | 10–15% | 1.9 (0.7–12) | Only 3% of KMT2Ar pediatric AML were M7 | tri 19, tri 21 | Low frequency of mutations, overexpression of HOX genes | Poor | [44,48,62,63,64] |
| t(9;11)(p22;q23) | KMT2A-MLLT3 (AF9) | 6–10% | ||||||
| t(10;11)(p12;q23)/ ins(10;11)(p12;q23q13) *** | KMT2A-MLLT10 (AF10) | 1–3% | ||||||
| t(6;11)(q27;q23) | KMT2A-MLLT4 (AF6) | 1% | ||||||
| t(11;17)(q23;q12) | KMT2A-MLLT6 (AF17) | 0.7–1% | ||||||
| t(11;19)(q23;p13.3) | KMT2A-MLLT1 (ENL) | 0.5–1% | ||||||
| t(4;11)(q21;q23) | KMT2A-AFF1 (AF4) | 0.5% | ||||||
| 12p13 abnormalities | NUP98-KMD5A ETV6 (12p13.1) del(12p) | Poor | [22,28] | |||||
| t(11;12)(p15;p13) * | NUP98-KMD5A | 10% | 1.9 (0.8–8.5) | 34% of cases were M7 | CK (numerous numerical and structural CA); RB1 deletion (13q14) | Low frequency of mutations; low RB1 expression; overexpression of HOX genes | Poor | [47,48,52,63,64] |
| t(7;12)(q36;p13) * | ETV6; MNX1 | very rare | 0.5 (0.2–1.9) | 4/42 cases were M7 Only infants | tri 19 (3/4 cases) | Unknown | Poor | [53] |
| HOX-r | HOX family genes (HOXA9, HOXA10, HOXB9, …) | 14% | trisomy 19, trisomy 21 | Overexpression of HOX genes | Good | [64,65] | ||
| t(3;7)(q21;p15.2) | GATA2-HOXA9 | rare | [64] | |||||
| t(3;7)(q21;p15.2) | GATA2-HOXA10 | rare | [64] | |||||
| t(5;7)(p13.2;p15.2) | NIPBL-HOXA9 | rare | [64] | |||||
| t(5;17)(p13.2;q21.3) | NIPBL-HOXB9 | rare | [64] | |||||
| t(11;22)(q24;q12) | MN1-FLI1 | rare | [141] | |||||
| GATA1 mutation | GATA1 (Xp11) truncating mutation | 7% | Search for a DS (mosaicism) | tri 21 in nearly all cases | Same gene expression profile as DS-AMKL | Good | [64] | |
| Monosomy 7 | / | 7–8% | 1.5 (0.5–17.1) | Exclude a primary abnormality and a predisposition syndrome (GATA2) | / | Frequently as part of a complex karyotype | Poor | [48,62,63,64] |
| Abnormal 7p | unknown (HOXA9?) | 12% | 1.8 (0.5–8.2) | 50% of abn7p cases were translocations; search for HOXr (7p15) | Good? Intermediate? | [62,64] | ||
| del13q | unknown (RB1?) | 4% | 1.5 (0.6–4.9) | Search for a primary stratifying CA that can be cryptic (NUP98-KDM5A) | [62] | |||
| Hyperdiploidy (47–84 chr) | % in AMKL: tri 21 (36%), tri 19 (24%) tri 8 (20%) tri 6 (15%) | 50% | Search for a primary stratifying CA that can be cryptic | / | / | According to cryptic CA and mutations or intermediate | [48,62,63] | |
| Hyperdiploidy (47–50 chr) | / | 38% | 1.7 (0.1–15) | Search for a primary stratifying CA that can be cryptic | / | / | [62] | |
| Hyperdiploidy (51–84 chr) | / | 12% | 1.7 (0.6–6.5) | Search for a primary stratifying CA that can be cryptic | / | / | [62] | |
| Complex | At least 3 independent CAs including a structural CA | 50% | 1.5 (0.4–15) | Search for a primary stratifying CA that can be cryptic | / | / | According to cryptic CA and mutations or intermediate | [5,6,22,28] |
| Normal karyotype | / | 13–16% | 1.5 (0.1–16) | Search for a cryptic CA or prognostic mutation | / | / | According to cryptic CA and mutations or intermediate | [48,62,63] |
| Risk Category | Pediatric AML Risk Stratification | Adult AML Risk Stratification (Excluding APL *) |
|---|---|---|
| Favorable | t(15;17)(q24;q21)/PML-RARA * t(8;21)(q22;q22)/RUNX1-RUNX1T1 inv(16)(p13q22) or t(16;16)(p13q22)/CBFB-MYH11 t(1;11)(q21;q23)/KMT2A-MLLT11(AF1Q) ** Cytogenetically normal cases with: -NPM1 mutation; - CEBPA double mutation GATA1 mutation **. | t(8;21)(q22;q22)/RUNX1-RUNX1T1 inv(16)(p13q22) or t(16;16)(p13q22)/CBFB-MYH11 NPM1 mutation without FLT3-ITD or with FLT3-ITDlow † CEBPA double mutation |
| Intermediate | CAs not classified as favorable or adverse | CAs not classified as favorable or adverse t(9;11)(p21;q23)/KMT2A-MLLT3 (AF9) ‡ NPM1 mutation with FLT3-ITDhigh † Wild-type NPM1 without FLT3-ITD or with FLT3-ITDlow † (without adverse-risk genetic lesions) |
| Adverse | inv(3)(q21q26) or t(3;3)(q21;q26)/GATA2; MECOM (EVI1) del(5q), -5 -7 ƒ t(6;9)(p23;q34)/DEK-NUP214 t(4;11)(q27;q23)/KMT2A-MLLT2(AF4) t(6;11)(q27;q23)/KMT2A-MLLT4(AF6) t(10;11)(p13;q23)/KMT2A-MLLT10(AF10) t(5;11)(q35;p13)/NUP98-NSD1 ** t(7;12)(q36;p13)/ETV6(TEL); HLXB9(MNX1) ** t(9;22)(q34;q11)/BCR-ABL1 Complex karyotype (≥3 CAs) ƒ FLT3-ITD mutation § WT1 mutation § | inv(3)(q21q26) or t(3;3)(q21;q26)/GATA2; MECOM (EVI1) del(5q), -5 -7 ƒ t(6;9)(p23;q34)/DEK-NUP214 t(v;11q23)/KMT2Ar †† t(9;22)(q34;q11)/BCR-ABL1 Complex karyotype (≥3 CAs) ƒ -17/abn17p and /or TP53 mutation # *** Monosomal karyotype ƒƒ FLT3-ITDhigh † § ASXL1 mutation § RUNX1 mutation § |
3. Special Considerations: FAB Subtype (M7), Age, Predisposition
3.1. Acute Megakaryoblastic Leukemia
3.2. Changes in Cytogenetic and Molecular Genetics According to Age
3.3. AML Predisposition Syndromes
4. Cytogenetics Versus Molecular Analysis
5. Conclusions/Prospective Considerations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Creutzig, U.; Heuvel-Eibrink, M.M.V.D.; Gibson, B.; Dworzak, M.N.; Adachi, S.; De Bont, E.; Harbott, J.; Hasle, H.; Johnston, D.; Kinoshita, A.; et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: Recommendations from an international expert panel. Blood 2012, 120, 3187–3205. [Google Scholar] [CrossRef] [PubMed]
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.; De Moerloose, B.; Dworzak, M.N.; Gibson, B.E.; et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Slats, A.M.; Egeler, R.M.; Berg, A.V.D.D.-V.D.; Korbijn, C.; Hählen, K.; Kamps, W.; Veerman, A.J.P.; Zwaan, C.M. Causes of death—Other than progressive leukemia—In childhood acute lymphoblastic (ALL) and myeloid leukemia (AML): The Dutch Childhood Oncology Group experience. Leukemia 2005, 19, 537–544. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.D.E.; Zwaan, C.M.; Van den Heuvel-Eibrink, M. Pediatric AML: From biology to clinical management. J. Clin. Med. 2015, 4, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Rasche, M.; Von Neuhoff, C.; Dworzak, M.; Bourquin, J.-P.; Bradtke, J.; Göhring, G.; Escherich, G.; Fleischhack, G.; Graf, N.; Gruhn, B.; et al. Genotype-outcome correlations in pediatric AML: The impact of a monosomal karyotype in trial AML-BFM 2004. Leukemia 2017, 31, 2807–2814. [Google Scholar] [CrossRef]
- Bager, N.; Juul-Dam, K.L.; Sandahl, J.D.; Abrahamsson, J.; Beverloo, B.; De Bont, E.S.J.M.; Ha, S.-Y.; Jahnukainen, K.; Ólafur, G.J.; Kaspers, G.L.; et al. Complex and monosomal karyotype are distinct cytogenetic entities with an adverse prognostic impact in paediatric acute myeloid leukaemia. A NOPHO-DBH-AML study. Br. J. Haematol. 2018, 183, 618–628. [Google Scholar] [CrossRef]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.; Choi, J.; Fisher, B.; Gerbing, R.; Hirsch, B.; Horton, T.; Kahwash, S.; et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: A report from the Children’s Oncology Group. Haematology 2020, 105, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Conneely, S.E.; Stevens, A.M. Acute myeloid leukemia in children: Emerging paradigms in genetics and new approaches to therapy. Curr. Oncol. Rep. 2021, 23, 1–13. [Google Scholar] [CrossRef]
- Roberts, I.; Izraeli, S. Haematopoietic development and leukaemia in Down syndrome. Br. J. Haematol. 2014, 167, 587–599. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Mecucci, C. Practical considerations for diagnosis and management of patients and carriers. Semin. Hematol. 2017, 54, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Catovsky, D.; Daniel, M.-T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the Acute Leukaemias French-American-British (FAB) Co-operative Group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposed revised criteria for the classification of acute myeloid leukemia. Ann. Intern. Med. 1985, 103, 620–625. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Harris, N.L.; Diebold, J.; Muller-Hermelink, H.K. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. A progress report. Am. J. Clin. Pathol. 1999, 111, S8–S12. [Google Scholar] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Rubnitz, J.; Inaba, H.; Dahl, G.; Ribeiro, R.C.; Bowman, W.P.; Taub, J.; Pounds, S.; Razzouk, B.; Lacayo, N.J.; Cao, X.; et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: Results of the AML02 multicentre trial. Lancet Oncol. 2010, 11, 543–552. [Google Scholar] [CrossRef]
- Laing, A.A.; Harrison, C.J.; Gibson, B.E.; Keeshan, K. Unlocking the potential of anti-CD33 therapy in adult and childhood acute myeloid leukemia. Exp. Hematol. 2017, 54, 40–50. [Google Scholar] [CrossRef]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III children’s oncology group trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Iland, H.J.; Collins, M.; Bradstock, K.; Supple, S.G.; Catalano, A.; Hertzberg, M.; Browett, P.; Grigg, A.; Firkin, F.; Campbell, L.J.; et al. Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: A non-randomised phase 2 trial. Lancet Haematol. 2015, 2, e357–e366. [Google Scholar] [CrossRef]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Hills, R.; Moorman, A.; Grimwade, D.J.; Hann, I.; Webb, D.K.H.; Wheatley, K.; De Graaf, S.S.N.; Berg, E.V.D.; Burnett, A.K.; et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment Trials AML 10 and 12. J. Clin. Oncol. 2010, 28, 2674–2681. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; O’Connor, C.; Cañete, A.; Bittencourt-Silvestre, J.; Sarrou, E.; Prendergast, Á.; Choi, J.; Johnston, P.; Wells, C.A.; Gibson, B.; et al. Age-specific biological and molecular profiling distinguishes paediatric from adult acute myeloid leukaemias. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Creutzig, U.; Zimmermann, M.; Reinhardt, D.; Rasche, M.; Von Neuhoff, C.; Alpermann, T.; Dworzak, M.; Perglerová, K.; Zemanova, Z.; Tchinda, J.; et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer 2016, 122, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Suzuki, M.; Otsuki, A.; Shimizu, R.; Bresnick, E.H.; Engel, J.D.; Yamamoto, M. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 2014, 25, 415–427. [Google Scholar] [CrossRef]
- Gröschel, S.; Sanders, M.A.; Hoogenboezem, R.; De Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; Van Der Velden, V.H.; Havermans, M.; Avellino, R.; Van Lom, K.; et al. A single oncogenic enhancer re-arrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef]
- Calvo, C.; Fenneteau, O.; Leverger, G.; Petit, A.; Baruchel, A.; Méchinaud, F. Infant acute myeloid leukemia: A unique clinical and biological entity. Cancers 2021, 13, 777. [Google Scholar] [CrossRef]
- Von Neuhoff, C.; Reinhardt, D.; Sander, A.; Zimmermann, M.; Bradtke, J.; Betts, D.R.; Zemanova, Z.; Stary, J.; Bourquin, J.P.; Haas, O.A.; et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J. Clin. Oncol. 2010, 28, 2682–2689. [Google Scholar] [CrossRef]
- Brown, J.; Jawad, M.; Twigg, S.R.F.; Saracoglu, K.; Sauerbrey, A.; Thomas, A.E.; Eils, R.; Harbott, J.; Kearney, L. A cryptic t(5;11)(q35;p15.5) in 2 children with acute myeloid leukemia with apparently normal karyotypes, identified by a multiplex fluorescence in situ hybridization telomere assay. Blood 2002, 99, 2526–2531. [Google Scholar] [CrossRef]
- Jaju, R.J.; Haas, O.; Neat, M.; Harbott, J.; Saha, V.; Boultwood, J.; Brown, J.M.; Pirc-Danoewinata, H.; Krings, B.W.; Müller, U.; et al. A new recurrent translocation, t(5;11)(q35;p15.5), associated with del(5q) in childhood acute myeloid leukemia. The UK Cancer Cytogenetics Group (UKCCG). Blood 1999, 94, 773–780. [Google Scholar]
- Rack, K.A.; Berg, E.V.D.; Haferlach, C.; Beverloo, H.B.; Costa, D.; Espinet, B.; Foot, N.; Jeffries, S.; Martin, K.; O’Connor, S.; et al. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia 2019, 33, 1851–1867. [Google Scholar] [CrossRef]
- Nguyen-Khac, F.; Bidet, A.; Veronese, L.; Daudignon, A.; Penther, D.; Troadec, M.-B.; Lefebvre, C.; Lafage-Pochitaloff, M. Recommendations for cytogenomic analysis of hematologic malignancies: Comments from the Francophone Group of Hematological Cytogenetics (GFCH). Leukemia 2019, 34, 1711–1713. [Google Scholar] [CrossRef]
- Rack, K.A.; Berg, E.V.D.; Haferlach, C.; Beverloo, H.B.; Costa, D.; Espinet, B.; Foot, N.; Jeffries, S.; Martin, K.; O’Connor, S.; et al. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms: Reponse to the comments from the Francophone Group of Hematological Cytogenetics (GFCH). Leukemia 2020, 34, 2262–2264. [Google Scholar] [CrossRef]
- Grimwade, D.; Biondi, A.; Mozziconacci, M.J.; Hagemeijer, A.; Berger, R.; Neat, M.; Howe, K.; Dastugue, N.; Jansen, J.; Radford-Weiss, I.; et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): Results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d’Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action “Molecular Cytogenetic Diagnosis in Haematological Malignancies”. Blood 2000, 96, 1297–1308. [Google Scholar] [PubMed]
- Vujkovic, M.; Attiyeh, E.F.; Ries, R.E.; Goodman, E.K.; Ding, Y.; Kavcic, M.; Alonzo, T.A.; Wang, Y.-C.; Gerbing, R.B.; Sung, L.; et al. Genomic architecture and treatment outcome in pediatric acute myeloid leukemia: A Children’s Oncology Group report. Blood 2017, 129, 3051–3058. [Google Scholar] [CrossRef] [PubMed]
- Marceau-Renaut, A.; Duployez, N.; Ducourneau, B.; Labopin, M.; Petit, A.; Rousseau, A.; Geffroy, S.; Bucci, M.; Cuccuini, W.; Fenneteau, O.; et al. Molecular profiling defines distinct prognostic subgroups in childhood AML: A report from the French ELAM02 Study Group. HemaSphere 2018, 2, e31. [Google Scholar] [CrossRef]
- Haferlach, C.; Rieder, H.; Lillington, D.M.; Dastugue, N.; Hagemeijer, A.; Harbott, J.; Stilgenbauer, S.; Knuutila, S.; Johansson, B.; Fonatsch, C.; et al. Proposals for standardized protocols for cytogenetic analyses of acute leukemias, chronic lymphocytic leukemia, chronic myeloid leukemia, chronic myeloproliferative disorders, and myelodysplastic syndromes. Genes Chromosom. Cancer 2007, 46, 494–499. [Google Scholar] [CrossRef]
- Luquet, I.; Bidet, A.; Cuccuini, W.; Lafage-Pochitaloff, M.; Mozziconacci, M.-J.; Terré, C. Place de la cytogénétique dans la prise en charge des leucémies aiguës myéloïdes: Actualisation par le Groupe francophone de cytogénétique hématologique (GFCH). Ann. Biol. Clin. 2016, 74, 12. [Google Scholar]
- Zhang, M.; Churpek, J.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet. 2015, 47, 180–185. [Google Scholar] [CrossRef]
- Klein, K.; Kaspers, G.; Harrison, C.J.; Beverloo, H.B.; Reedijk, A.; Bongers, M.; Cloos, J.; Pession, A.; Reinhardt, D.; Zimmerman, M.; et al. Clinical impact of additional cytogenetic aberrations, cKIT and RAS mutations, and treatment elements in pediatric t(8;21)-AML: Results from an international retrospective study by the International Berlin-Frankfurt-Münster Study Group. J. Clin. Oncol. 2015, 33, 4247–4258. [Google Scholar] [CrossRef] [PubMed]
- Faber, Z.; Chen, X.; Gedman, A.L.; Boggs, K.; Cheng, J.; Ma, J.; Radtke, I.; Chao, J.-R.; Walsh, M.P.; Song, G.; et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 2016, 48, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Marceau-Renaut, A.; Boissel, N.; Petit, A.; Bucci, M.; Geffroy, S.; Lapillonne, H.; Renneville, A.; Ragu, C.; Figeac, M.; et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 2016, 127, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Boudry-Labis, E.; Roumier, C.; Boissel, N.; Petit, A.; Geffroy, S.; Helevaut, N.; Celli-Lebras, K.; Terré, C.; Fenneteau, O.; et al. SNP-array lesions in core binding factor acute myeloid leukemia. Oncotarget 2018, 9, 6478–6489. [Google Scholar] [CrossRef]
- Balgobind, B.V.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: Results of an international retrospective study. Blood 2009, 114, 2489–2496. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Struski, S.; Lagarde, S.; Bories, P.; Puiseux, C.; Prade, N.; Cuccuini, W.; Pages, M.-P.; Bidet, A.; Gervais, C.; Lafage-Pochitaloff, M.; et al. NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 2016, 31, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Shiba, N.; Ohki, K.; Tabuchi, K.; Yamato, G.; Park, M.-J.; Tomizawa, D.; Kinoshita, A.; Shimada, A.; Arakawa, H.; et al. Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosom. Cancer 2017, 56, 394–404. [Google Scholar] [CrossRef]
- Niktoreh, N.; Walter, C.; Zimmermann, M.; Von Neuhoff, C.; Von Neuhoff, N.; Rasche, M.; Waack, K.; Creutzig, U.; Hanenberg, H.; Reinhardt, D. MutatedWT1, FLT3-ITD, and NUP98-NSD1 Fusion in various combinations define a poor prognostic group in pediatric acute myeloid leukemia. J. Oncol. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shiba, N.; Yoshida, K.; Hara, Y.; Yamato, G.; Shiraishi, Y.; Matsuo, H.; Okuno, Y.; Chiba, K.; Tanaka, H.; Kaburagi, T.; et al. Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv. 2019, 3, 3157–3169. [Google Scholar] [CrossRef] [PubMed]
- Hollink, I.H.I.M.; Heuvel-Eibrink, M.M.V.D.; Arentsen-Peters, S.T.C.J.M.; Pratcorona, M.; Abbas, S.; Kuipers, J.E.; Van Galen, J.F.; Beverloo, H.B.; Sonneveld, E.; Kaspers, G.-J.J.L.; et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011, 118, 3645–3656. [Google Scholar] [CrossRef]
- De Rooij, J.D.; Hollink, I.H.; Arentsen-Peters, S.T.C.J.M.; Van Galen, J.F.; Beverloo, H.B.; Baruchel, A.; Trka, J.; Reinhardt, D.; Sonneveld, E.; Zimmermann, M.; et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 2013, 27, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- Espersen, A.D.L.; Noren-Nyström, U.; Abrahamsson, J.; Ha, S.-Y.; Pronk, C.J.; Jahnukainen, K.; Jónsson Ólafur, G.; Lausen, B.; Palle, J.; Zeller, B.; et al. Acute myeloid leukemia (AML) with t(7;12)(q36;p13) is associated with infancy and trisomy 19: Data from Nordic Society for Pediatric Hematology and Oncology (NOPHO-AML) and review of the literature. Genes Chromosom. Cancer 2018, 57, 359–365. [Google Scholar] [CrossRef]
- Borel, C.; Dastugue, N.; Cances-Lauwers, V.; Mozziconacci, M.-J.; Prebet, T.; Vey, N.; Pigneux, A.; Lippert, E.; Visanica, S.; Legrand, F.; et al. PICALM–MLLT10 acute myeloid leukemia: A French cohort of 18 patients. Leuk. Res. 2012, 36, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.; Choi, J.R.; Kim, M.J.; Kim, S.Y.; Lee, H.J.; Suh, J.T.; Yoon, H.J.; Lee, J.; Lee, S.; Lee, W.I.; et al. Detection of t(3;5) and NPM1/MLF1 rearrangement in an elderly patient with acute myeloid leukemia: Clinical and laboratory study with review of the literature. Cancer Genet. Cytogenet. 2010, 199, 101–229. [Google Scholar] [CrossRef] [PubMed]
- Sandahl, J.D.; Kjeldsen, E.; Abrahamsson, J.; Ha, S.-Y.; Heldrup, J.; Jahnukainen, K.; Jónsson Ólafur, G.; Lausen, B.; Palle, J.; Zeller, B.; et al. Ploidy and clinical characteristics of childhood acute myeloid leukemia: A NOPHO-AML study. Genes Chromosom. Cancer 2014, 53, 667–675. [Google Scholar] [CrossRef]
- Tarlock, K.; Alonzo, T.A.; Moraleda, P.P.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Ravindranath, Y.; Lange, B.; Woods, W.G.; Gamis, A.S.; et al. Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless ofFLT3-ITD status: A report from the Children’s Oncology Group. Br. J. Haematol. 2014, 166, 254–259. [Google Scholar] [CrossRef]
- Coenen, E.A.; Zwaan, C.M.; Reinhardt, D.; Harrison, C.J.; Haas, O.A.; De Haas, V.; Mihál, V.; De Moerloose, B.; Jeison, M.; Rubnitz, J.E.; et al. Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity: A collaborative study by the International-Berlin-Frankfurt-Münster AML-study group. Blood 2013, 122, 2704–2713. [Google Scholar] [CrossRef]
- Noort, S.; Zimmermann, M.; Reinhardt, D.; Cuccuini, W.; Pigazzi, M.; Smith, J.; Ries, R.E.; Alonzo, T.A.; Hirsch, B.; Tomizawa, D.; et al. Prognostic impact of t(16;21)(p11;q22) and t(16;21)(q24;q22) in pediatric AML: A retrospective study by the I-BFM Study Group. Blood 2018, 132, 1584–1592. [Google Scholar] [CrossRef]
- Bernstein, J.; Dastugue, N.; Haas, O.; Harbott, J.; Heerema, N.; Huret, J.L.; Landman-Parker, J.; Lebeau, M.M.; Leonard, C.; Mann, G.; et al. Nineteen cases of the t(1;22)(p13;q13) acute megakaryblastic leukaemia of infants/children and a review of 39 cases: Report from a t(1;22) study group. Leukemia 2000, 14, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Dastugue, N.; Lafage-Pochitaloff, M.; Pagès, M.-P.; Radford, I.; Bastard, C.; Talmant, P.; Mozziconacci, M.J.; Leonard, C.; Bilhou-Nabera, C.; Cabrol, C.; et al. Cytogenetic profile of childhood and adult megakaryoblastic leukemia (M7): A study of the Groupe Francais de Cytogenetique Hematologique (GFCH). Blood 2002, 100, 618–626. [Google Scholar] [CrossRef]
- Inaba, H.; Zhou, Y.; Abla, O.; Adachi, S.; Auvrignon, A.; Beverloo, H.B.; De Bont, E.; Chang, T.-T.; Creutzig, U.; Dworzak, M.N.; et al. Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: A retrospective international study. Blood 2015, 126, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.D.; Branstetter, C.; Ma, J.; Li, Y.; Walsh, M.P.; Cheng, J.; Obulkasim, A.; Dang, J.; Easton, J.; Verboon, L.J.; et al. Pediatric non–Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017, 49, 451–456. [Google Scholar] [CrossRef]
- De Rooij, J.D.; Masetti, R.; Van Den Heuvel-Eibrink, M.M.; Cayuela, J.M.; Trka, J.; Reinhardt, D.; Rasche, M.; Sonneveld, E.; Alonzo, T.A.; Fornerod, M.; et al. Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: A retrospective intergroup study. Blood 2016, 127, 3424–3430. [Google Scholar] [CrossRef]
- Masetti, R.; Bertuccio, S.N.; Pession, A.; Locatelli, F. CBFA2T3-GLIS2-positive acute myeloid leukaemia. A peculiar paediatric entity. Br. J. Haematol. 2018, 184, 337–347. [Google Scholar] [CrossRef]
- Masetti, R.; Pigazzi, M.; Togni, M.; Astolfi, A.; Indio, V.; Manara, E.; Casadio, R.; Pession, A.; Basso, G.; Locatelli, F. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 2013, 121, 3469–3472. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Ries, R.E.; Hylkema, T.; Alonzo, T.A.; Gerbing, R.B.; Santaguida, M.T.; Brodersen, L.E.; Pardo, L.; Cummings, C.L.; Loeb, K.R.; et al. Comprehensive transcriptome profiling of cryptic CBFA2T3–GLIS2 fusion–positive AML defines novel therapeutic options: A COG and TARGET pediatric AML study. Clin. Cancer Res. 2019, 26, 726–737. [Google Scholar] [CrossRef]
- Johnston, D.L.; Alonzo, T.A.; Gerbing, R.B.; Hirsch, B.; Heerema, N.A.; Ravindranath, Y.; Woods, W.G.; Lange, B.J.; Gamis, A.S.; Raimondi, S.C. Outcome of pediatric patients with acute myeloid leukemia (AML) and -5/5q- abnormalities from five pediatric AML treatment protocols: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2013, 60, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Hasle, H.; Alonzo, T.A.; Auvrignon, A.; Behar, C.; Chang, M.; Creutzig, U.; Fischer, A.; Forestier, E.; Fynn, A.; Haas, O.A.; et al. Monosomy 7 and deletion 7q in children and ado-lescents with acute myeloid leukemia: An international retrospective study. Blood 2007, 109, 4641–4647. [Google Scholar] [CrossRef]
- Laursen, A.C.L.; Sandahl, J.D.; Kjeldsen, E.; Abrahamsson, J.; Asdahl, P.; Ha, S.-Y.; Heldrup, J.; Jahnukainen, K.; Jónsson Ólafur, G.; Lausen, B.; et al. Trisomy 8 in pediatric acute myeloid leukemia: A NOPHO-AML study. Genes Chromosom. Cancer 2016, 55, 719–726. [Google Scholar] [CrossRef]
- Chilton, L.; Hills, R.; Harrison, C.J.; Burnett, A.K.; Grimwade, D.; Moorman, A.V. Hyperdiploidy with 49–65 chromosomes represents a heterogeneous cytogenetic subgroup of acute myeloid leukemia with differential outcome. Leukemia 2014, 28, 321–328. [Google Scholar] [CrossRef]
- Gregory, J.; Feusner, J. Acute promyelocytic leukemia in childhood. Curr. Oncol. Rep. 2009, 11, 439–445. [Google Scholar] [CrossRef]
- Zhang, L.; Samad, A.; Pombo-De-Oliveira, M.; Scelo, G.; Smith, M.; Feusner, J.; Wiemels, J.; Metayer, C. Global characteristics of childhood acute promyelocytic leukemia. Blood Rev. 2015, 29, 101–125. [Google Scholar] [CrossRef]
- Sandoval, C.; Pui, C.H.; Bowman, L.C.; Heaton, D.; Hurwitz, C.; Raimondi, S.C.; Behm, F.G.; Head, D.R. Secondary acute myeloid leukemia in children previously treated with alkylating agents, intercalating topoisomerase II inhibitors, and irradiation. J. Clin. Oncol. 1993, 11, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- De Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef]
- Gregory, J.; Kim, H.; Alonzo, T.; Gerbing, R.; Woods, W.; Weinstein, H.; Shepherd, L.; Schiffer, C.; Appelbaum, F.; Willman, C.; et al. Treatment of children with acute promyelocytic leu-kemia: Results of the first North American Intergroup trial INT0129. Pediatr. Blood Cancer 2009, 53, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Conneely, S.; Stevens, A. Advances in pediatric acute promyelocytic leukemia. Child 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Labrador, J.; Luño, E.; Vellenga, E.; Brunet, S.; González-Campos, J.; Chillón, M.C.; Holowiecka, A.; Esteve, J.; Bergua, J.; González-Sanmiguel, J.D.; et al. Clinical significance of complex karyotype at diagnosis in pediatric and adult patients with de novo acute promyelocytic leukemia treated with ATRA and chemotherapy. Leuk. Lymphoma 2018, 60, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Poiré, X.; Moser, B.K.; Gallagher, R.E.; Laumann, K.; Bloomfield, C.D.; Powell, B.L.; Koval, G.; Gulati, K.; Holowka, N.; Larson, R.A.; et al. Arsenic trioxide in front-line therapy of acute promyelocytic leukemia (C9710): Prognostic significance of FLT3 mutations and complex karyotype. Leuk. Lymphoma 2014, 55, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.; Grimwade, D. Prognostic significance of additional cytogenetic abnormalities and FLT3 mutations in acute pro-myelocytic leukemia. Leuk Lymphoma 2014, 55, 1444–1446. [Google Scholar] [CrossRef][Green Version]
- Sainty, D.; Liso, V.; Cantù-Rajnoldi, A.; Head, D.; Mozziconacci, M.J.; Arnoulet, C.; Benattar, L.; Fenu, S.; Mancini, M.; Duchayne, E.; et al. A new morphologic classification system for acute promyelocytic leukemia distinguishes cases with underlying PLZF/RARA gene rearrangements. Blood 2000, 96, 1287–1296. [Google Scholar] [PubMed]
- Picharski, G.L.; Andrade, D.P.; Fabro, A.L.M.R.; Lenzi, L.; Tonin, F.S.; Ribeiro, R.C.; Figueiredo, B.C.; Lenzi, L. The impact of Flt3 gene mutations in acute promyelocytic leukemia: A meta-analysis. Cancers 2019, 11, 1311. [Google Scholar] [CrossRef]
- Paschka, P.; Döhner, K. Core-binding factor acute myeloid leukemia: Can we improve on HiDAC consolidation? Hematology 2013, 2013, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Castilla, L.H.; Garrett, L.; Adya, N.; Orlic, D.; Dutra, A.; Anderson, S.; Owens, J.; Eckhaus, M.; Bodine, D.; Liu, P.P. The fusion gene Cbfb-MYH11 blocks myeloid differ-entiation and predisposes mice to acute myelomonocytic leukaemia. Nat. Genet. 1999, 23, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; O’Brien, D.; Kumaravelu, P.; Lenny, N.; Yeoh, E.-J.; Downing, J.R. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 2002, 1, 63–74. [Google Scholar] [CrossRef]
- Kühn, M.W.M.; Radtke, I.; Bullinger, L.; Goorha, S.; Cheng, J.; Edelmann, J.; Gohlke, J.; Su, X.; Paschka, P.; Pounds, S.; et al. High-resolution genomic profiling of adult and pe-diatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 2012, 119, e67–e75. [Google Scholar] [CrossRef]
- Grimwade, D.; Freeman, S.D. Defining minimal residual disease in acute myeloid leukemia: Which platforms are ready for “prime time”? Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 222–233. [Google Scholar] [CrossRef]
- Gamerdinger, U.; Teigler-Schlegel, A.; Pils, S.; Bruch, J.; Viehmann, S.; Keller, M.; Jauch, A.; Harbott, J. Cryptic chromosomal aberrations leading to anAML1/ETO rearrangement are frequently caused by small insertions. Genes Chromosom. Cancer 2003, 36, 261–272. [Google Scholar] [CrossRef]
- Jahn, N.; Terzer, T.; Sträng, E.; Dolnik, A.; Cocciardi, S.; Panina, E.; Corbacioglu, A.; Herzig, J.; Weber, D.; Schrade, A.; et al. Genomic heterogeneity in core-binding factor acute myeloid leukemia and its clinical implication. Blood Adv. 2020, 4, 6342–6352. [Google Scholar] [CrossRef]
- Paschka, P.; Du, J.; Schlenk, R.F.; Gaidzik, V.I.; Bullinger, L.; Corbacioglu, A.; Späth, D.; Kayser, S.; Schlegelberger, B.; Krauter, J.; et al. Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): A study of the German-Austrian AML Study Group (AMLSG). Blood 2013, 121, 170–177. [Google Scholar] [CrossRef]
- Bindels, E.M.; Havermans, M.; Lugthart, S.; Erpelinck, C.; Wocjtowicz, E.; Krivtsov, A.V.; Rombouts, E.; Armstrong, S.A.; Taskesen, E.; Haanstra, J.R.; et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood 2012, 119, 5838–5849. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.F.; Sukov, W.R.; Pitel, B.A.; Smoley, S.A.; Pearce, K.E.; Meyer, R.G.; Williamson, C.M.; Smadbeck, J.B.; Vasmatzis, G.; Hoppman, N.L.; et al. Acute leukemias harboring KMT2A/MLLT10 fusion: A 10-year experience from a single genomics laboratory. Genes Chromosom. Cancer 2019, 58, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Barber, K.E.; Ford, A.M.; Harris, R.L.; Harrison, C.J.; Moorman, A.V. MLL translocations with concurrent 3? deletions: Interpretation of FISH results. Genes Chromosom. Cancer 2004, 41, 266–271. [Google Scholar] [CrossRef]
- Fu, J.-F.; Hsu, J.-J.; Tang, T.-C.; Shih, L.-Y. Identification of CBL, a proto-oncogene at 11q23.3, as a novel MLL fusion partner in a patient with de novo acute myeloid leukemia. Genes Chromosom. Cancer 2003, 37, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Ostronoff, F.; Othus, M.; Gerbing, R.B.; Loken, M.R.; Raimondi, S.C.; Hirsch, B.A.; Lange, B.J.; Petersdorf, S.; Radich, J.; Appelbaum, F.R.; et al. NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: A COG and SWOG report. Blood 2014, 124, 2400–2407. [Google Scholar] [CrossRef]
- Hara, Y.; Shiba, N.; Yamato, G.; Ohki, K.; Tabuchi, K.; Sotomatsu, M.; Tomizawa, D.; Kinoshita, A.; Arakawa, H.; Saito, A.M.; et al. Patients aged less than 3 years with acute myeloid leukaemia characterize a molecularly and clinically distinct subgroup. Br. J. Haematol. 2020, 188, 528–539. [Google Scholar] [CrossRef]
- McNeer, N.A.; Philip, J.; Geiger, H.; Ries, R.E.; Lavallée, V.-P.; Walsh, M.; Shah, M.; Arora, K.; Emde, A.-K.; Robine, N.; et al. Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 2019, 33, 1934–1943. [Google Scholar] [CrossRef]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef]
- Noort, S.; Wander, P.; Alonzo, T.A.; Smith, J.; Ries, R.E.; Gerbing, R.B.; Dolman, M.E.M.; Locatelli, F.; Reinhardt, D.; Baruchel, A.; et al. The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematology 2020, 106, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, K.M.; Heerema-McKenney, A.E.; Choi, J.K.; Smith, J.; Ries, R.E.; Hirsch, B.A.; Raimondi, S.C.; Alonzo, T.A.; Wang, Y.-C.; Aplenc, R.; et al. Acute erythroid leukemia is enriched in NUP98 fusions: A report from the Children’s Oncology Group. Blood Adv. 2020, 4, 6000–6008. [Google Scholar] [CrossRef] [PubMed]
- Nebral, K.; König, M.; Schmidt, H.H.; Lutz, D.; Sperr, W.R.; Kalwak, K.; Brugger, S.; Dworzak, M.N.; Haas, O.; Strehl, S. Screening for NUP98 rearrangements in hematopoietic malignancies by fluorescence in situ hybridization. Haematology 2005, 90, 746–752. [Google Scholar]
- Ballabio, E.; Cantarella, C.D.; Federico, C.; Di Mare, P.; Hall, G.; Harbott, J.; Hughes, J.; Saccone, S.; Tosi, S. Ectopic expression of the HLXB9 gene is associated with an altered nuclear position in t(7;12) leukaemias. Leukemia 2009, 23, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Ingenhag, D.; Reister, S.; Auer, F.; Bhatia, S.; Wildenhain, S.; Picard, D.; Remke, M.; Hoell, J.I.; Kloetgen, A.; Sohn, D.; et al. The homeobox transcription factor HB9 induces senescence and blocks differentiation in hematopoietic stem and progenitor cells. Haematology 2018, 104, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Naiel, A.; Vetter, M.; Plekhanova, O.; Fleischman, E.; Sokova, O.; Tsaur, G.; Harbott, J.; Tosi, S. A Novel three-colour fluorescence in situ hybridization approach for the detection of t(7;12)(q36;p13) in acute myeloid leukaemia reveals new cryptic three way translocation t(7;12;16). Cancers 2013, 5, 281–295. [Google Scholar] [CrossRef]
- Tosi, S.; Kamel, Y.M.; Owoka, T.; Federico, C.; Truong, T.H.; Saccone, S. Paediatric acute myeloid leukaemia with the t(7;12)(q36;p13) rearrangement: A review of the biological and clinical management aspects. Biomark. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef]
- Lugthart, S.; Gröschel, S.; Beverloo, H.B.; Kayser, S.; Valk, P.J.M.; Van Zelderen-Bhola, S.L.; Ossenkoppele, G.J.; Vellenga, E.; Ruiter, E.V.D.B.-D.; Schanz, U.; et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 3890–3898. [Google Scholar] [CrossRef]
- Ladigan, S.; Mika, T.; Figge, A.; May, A.M.; Schmiegel, W.; Schroers, R.; Baraniskin, A. Acute myeloid leukemia with central diabetes insipidus. Blood Cells, Mol. Dis. 2019, 76, 45–52. [Google Scholar] [CrossRef]
- Sandahl, J.D.; Coenen, E.A.; Forestier, E.; Harbott, J.; Johansson, B.; Kerndrup, G.; Adachi, S.; Auvrignon, A.; Beverloo, H.B.; Cayuela, J.-M.; et al. t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: An international study of 62 patients. Haematology 2014, 99, 865–872. [Google Scholar] [CrossRef]
- Von Lindern, M.; Fornerod, M.; Van Baal, S.; Jaegle, M.; De Wit, T.; Buijs, A.; Grosveld, G. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol. Cell. Biol. 1992, 12, 1687–1697. [Google Scholar] [CrossRef]
- Raimondi, S.C.; Dubé, I.D.; Valentine, M.B.; Mirro, J.; Watt, H.J.; Larson, R.; Bitter, M.; Le Beau, M.M.; Rowley, J.D. Clinicopathologic manifestations and breakpoints of the t(3;5) in patients with acute nonlymphocytic leukemia. Leukemia 1989, 3, 42–47. [Google Scholar]
- Grimwade, D.; Hills, R.; Moorman, A.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K.; on behalf of the National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Bolli, N.; Martelli, M.P.; Liso, A.; Gorello, P.; Mandelli, F.; Mecucci, C. Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematology 2007, 92, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Yoneda-Kato, N.; Look, A.T.; Kirstein, M.N.; Valentine, M.B.; Raimondi, S.C.; Cohen, K.J.; Carroll, A.J.; Morris, S.W. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene 1996, 12, 265–275. [Google Scholar]
- Borrow, J.; Stanton, V.P.; Andresen, J.M.; Becher, R.; Behm, F.G.; Chaganti, R.S.K.; Civin, C.I.; Disteche, C.; Dubé, I.; Frischauf, A.M.; et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB–binding protein. Nat. Genet. 1996, 14, 33–41. [Google Scholar] [CrossRef]
- Roberts, I.; Fordham, N.J.; Rao, A.; Bain, B.J. Neonatal leukaemia. Br. J. Haematol. 2018, 182, 170–184. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Åman, P.; Fioretos, T.; Höglund, M.; Johansson, B.; Mandahl, N.; Heim, S.; Behrendtz, M.; Mitelman, F. Fusion of theFUS gene withERG in acute myeloid leukemia with t(16;21)(p11;q22). Genes Chromosom. Cancer 1994, 11, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.T.; Ida, K.; Ichikawa, H.; Shimizu, K.; Ohki, M.; Maseki, N.; Kaneko, Y.; Sako, M.; Kobayashi, Y.; Tojou, A.; et al. Consistent detection of TLS/FUS-ERG chimeric transcripts in acute myeloid leukemia with t(16;21)(p11;q22) and identification of a novel transcript. Blood 1997, 90, 1192–1199. [Google Scholar]
- Gamou, T.; Kitamura, E.; Hosoda, F.; Shimizu, K.; Shinohara, K.; Hayashi, Y.; Nagase, T.; Yokoyama, Y.; Ohki, M. The partner gene of AML1 in t(16;21) myeloid malignancies is a novel member of the MTG8(ETO) family. Blood 1998, 91, 4028–4037. [Google Scholar] [CrossRef] [PubMed]
- Mercher, T.; Raffel, G.D.; Moore, S.A.; Cornejo, M.G.; Baudry-Bluteau, D.; Cagnard, N.; Jesneck, J.L.; Pikman, Y.; Cullen, D.; Williams, I.R.; et al. The OTT-MAL fusion oncogene activates RBPJ-mediated transcription and induces acute megakaryoblastic leukemia in a knockin mouse model. J. Clin. Investig. 2009, 119, 852–864. [Google Scholar] [CrossRef]
- Ma, Z.; Morris, S.W.; Valentine, V.; Martin, L.; Herbrick, J.-A.; Cui, X.; Bouman, D.; Li, Y.; Mehta, P.K.; Nizetic, D.; et al. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat. Genet. 2001, 28, 220–221. [Google Scholar] [CrossRef]
- Thiollier, C.; Pflumio, F.; Ballerini, P.; Crispino, J.D.; Bernard, O.; Mercher, T. Novel ETO2-GLIS2 fusion and therapeutic strategy in acute megakaryoblastic leukemia. Med. Sci. 2012, 28, 1013–1016. [Google Scholar]
- Gruber, T.A.; Gedman, A.L.; Zhang, J.; Koss, C.S.; Marada, S.; Ta, H.Q.; Chen, S.-C.; Su, X.; Ogden, S.K.; Dang, J.; et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 Fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 2012, 22, 683–697. [Google Scholar] [CrossRef]
- Lopez, C.K.; Noguera, E.; Stavropoulou, V.; Robert, E.; Aid, Z.; Ballerini, P.; Bilhou-Nabera, C.; Lapillonne, H.; Boudia, F.; Thirant, C.; et al. Ontogenic changes in hematopoietic hierarchy determine pediatric specificity and disease phenotype in fusion oncogene–driven myeloid leukemia. Cancer Discov. 2019, 9, 1736–1753. [Google Scholar] [CrossRef]
- Pardo, L.M.; Voigt, A.; Alonzo, T.A.; Wilson, E.R.; Gerbing, R.B.; Paine, D.J.; Dai, F.; Menssen, A.J.; Raimondi, S.C.; Hirsch, B.A.; et al. Deciphering the significance of CD56 expression in pediatric acute myeloid leukemia: A report from the Children’s Oncology Group. Cytom. B Clin. Cytom. 2020, 98, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Hasle, H.; for the European Working Group on MDS in Childhood (EWOG-MDS); Arico, M.; Basso, G.; Biondi, A.; Rajnoldi, A.C.; Creutzig, U.; Fenu, S.; Fonatsch, C.; Haas, O.; et al. Myelodysplastic syndrome, juvenile myelomonocytic leukemia, and acute myeloid leukemia associated with complete or partial monosomy 7. Leukemia 1999, 13, 376–385. [Google Scholar] [CrossRef]
- Davidsson, J.; Puschmann, A.; Tedgård, U.; Bryder, D.; Nilsson, L.; Cammenga, J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 2018, 32, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Collin, M.; Horwitz, M.S. GATA2 deficiency and related myeloid neoplasms. Semin. Hematol. 2017, 54, 81–86. [Google Scholar] [CrossRef]
- Paulsson, K.; Békássy, A.; Olofsson, T.; Mitelman, F.; Johansson, B.; Panagopoulos, I. A novel and cytogenetically cryptic t(7;21)(p22;q22) in acute myeloid leukemia results in fusion of RUNX1 with the ubiquitin-specific protease gene USP42. Leukemia 2005, 20, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Paulraj, P.; Diamond, S.; Razzaqi, F.; Ozeran, J.D.; Longhurst, M.; Andersen, E.F.; Toydemir, R.M.; Hong, B.; Ozeran, D. Pediatric acute myeloid leukemia with t(7;21)(p22;q22). Genes Chromosom. Cancer 2019, 58, 551–557. [Google Scholar] [CrossRef]
- Breems, D.A.; Van Putten, W.L.J.; De Greef, G.E.; Van Zelderen-Bhola, S.L.; Gerssen-Schoorl, K.B.J.; Mellink, C.H.M.; Nieuwint, A.; Jotterand, M.; Hagemeijer, A.; Beverloo, H.B.; et al. Monosomal karyotype in acute myeloid leukemia: A better indicator of poor prognosis than a complex karyotype. J. Clin. Oncol. 2008, 26, 4791–4797. [Google Scholar] [CrossRef]
- Luquet, I.; Laï, J.L.; Barin, C.; Baranger, L.; Bilhou-Nabera, C.; Lippert, E.; Gervais, C.; Talmant, P.; Cornillet-Lefebvre, P.; Pérot, C.; et al. Hyperdiploid karyotypes in acute myeloid leukemia define a novel entity: A study of 38 patients from the Groupe Francophone de Cytogenetique Hematologique (GFCH). Leukemia 2007, 22, 132–137. [Google Scholar] [CrossRef]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Meshinchi, S.; Arceci, R.J. Prognostic factors and risk-based therapy in pediatric acute myeloid leukemia. Oncologist 2007, 12, 341–355. [Google Scholar] [CrossRef]
- Meshinchi, S.; Arceci, R.J.; Sanders, J.E.; Smith, F.O.; Woods, W.B.; Radich, J.P.; Alonzo, T.A. Role of allogeneic stem cell transplantation in FLT3/ITD-positive AML. Blood 2006, 108, 400–401. [Google Scholar] [CrossRef]
- Tarlock, K.; Alonzo, T.; Gerbing, R.B.; Ries, R.E.; Gibson, B.; Niktoreh, N.; Noort, S.; Heuvel-Eibrink, M.M.V.D.; Zwaan, C.M.; Meshinchi, S. Distinct co-occurring mutational profiles in acute myeloid leukemia confers prognostic significance in children and young adults with FLT3/ITD mutations. Blood 2018, 132, 443. [Google Scholar] [CrossRef]
- Sahoo, R.K.; Kumar, L.; Eskazan, A.E.; Stone, R.M.; Larson, R.; Dohner, H. Midostaurin in FLT3-mutated acute myeloid leukemia. N. Engl. J. Med. 2017, 377, 1901–1903. [Google Scholar] [CrossRef] [PubMed]
- Hollink, I.H.I.M.; Zwaan, C.M.; Zimmermann, M.; Arentsen-Peters, T.C.J.M.; Pieters, R.; Cloos, J.; Kaspers, G.J.L.; De Graaf, S.S.N.; Harbott, J.; Creutzig, U.; et al. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 2008, 23, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; McIntyre, E.; Rau, R.; Meshinchi, S.; Lacayo, N.; Dahl, G.; Alonzo, T.A.; Chang, M.; Arceci, R.J.; Small, N. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 2007, 110, 979–985. [Google Scholar] [CrossRef]
- Ho, P.A.; Alonzo, T.A.; Gerbing, R.B.; Pollard, J.; Stirewalt, D.L.; Hurwitz, C.; Heerema, N.A.; Hirsch, B.; Raimondi, S.C.; Lange, B.; et al. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): A report from the Children’s Oncology Group. Blood 2009, 113, 6558–6566. [Google Scholar] [CrossRef]
- Rio-Machin, A.; Vulliamy, T.; Hug, N.; Walne, A.; Tawana, K.; Cardoso, S.; Ellison, A.; Pontikos, N.; Wang, J.; Tummala, H.; et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.A.; Downing, J.R. The biology of pediatric acute megakaryoblastic leukemia. Blood 2015, 126, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Labuhn, M.; Perkins, K.; Matzk, S.; Varghese, L.; Garnett, C.; Papaemmanuil, E.; Metzner, M.; Kennedy, A.; Amstislavskiy, V.; Risch, T.; et al. Mechanisms of progression of myeloid preleukemia to transformed myeloid leukemia in children with Down syndrome. Cancer Cell 2019, 36, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Taub, J.W.; Berman, J.N.; Hitzler, J.K.; Sorrell, A.D.; Lacayo, N.J.; Mast, K.; Head, D.; Raimondi, S.; Hirsch, B.; Ge, Y.; et al. Improved outcomes for myeloid leukemia of Down syndrome: A report from the Children’s Oncology Group AAML0431 trial. Blood 2017, 129, 3304–3313. [Google Scholar] [CrossRef]
- Uffmann, M.; Rasche, M.; Zimmermann, M.; Von Neuhoff, C.; Creutzig, U.; Dworzak, M.; Scheffers, L.; Hasle, H.; Zwaan, C.M.; Reinhardt, D.; et al. Therapy reduction in patients with Down syndrome and myeloid leukemia: The international ML-DS 2006 trial. Blood 2017, 129, 3314–3321. [Google Scholar] [CrossRef]
- Athale, U.H.; Razzouk, B.I.; Raimondi, S.C.; Tong, X.; Behm, F.G.; Head, D.R.; Srivastava, D.K.; Rubnitz, J.E.; Bowman, L.; Pui, C.H.; et al. Biology and outcome of childhood acute megakaryoblastic leukemia: A single institution’s experience. Blood 2001, 97, 3727–3732. [Google Scholar] [CrossRef]
- Pagano, L.; Gimema, F.T.; Pulsoni, A.; Vignetti, M.; Mele, L.; Fianchi, L.; Petti, M.; Mirto, S.; Falcucci, P.; Fazi, P.; et al. Acute megakaryoblastic leukemia: Experience of GIMEMA trials. Leukemia 2002, 16, 1622–1626. [Google Scholar] [CrossRef]
- Prudowsky, Z.; Han, H.; Stevens, A. Transient abnormal myelopoeisis and mosaic down syndrome in a phenotypically normal newborn. Child 2020, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Vendemini, F.; Zama, D.; Biagi, C.; Pession, A.; Locatelli, F. Acute Myeloid leukemia in infants: Biology and treatment. Front. Pediatr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M.; Flotho, C. Juvenile myelomonocytic leukemia: Who’s the driver at the wheel? Blood 2019, 133, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.D.; Lindsley, R.C. Clonal hematopoiesis in the inherited bone marrow failure syndromes. Blood 2020, 136, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Cavenagh, J.D.; Lister, T.A.; Fitzgibbon, J. Mutation of CEBPA in familial acute myeloid leukemia. N. Engl. J. Med. 2004, 351, 2403–2407. [Google Scholar] [CrossRef]
- Latger-Cannard, V.; Philippe, C.; Bouquet, A.; Baccini, V.; Alessi, M.-C.; Ankri, A.; Bauters, A.; Bayart, S.; Cornillet-Lefebvre, P.; Daliphard, S.; et al. Haematological spectrum and genotype-phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet J. Rare Dis. 2016, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; Van Den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Favier, R.; Alessi, M.-C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M.; et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood 2013, 122, 1987–1989. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Hörstedt, A.S.; Johansson, B.; Antunovic, P.; Billström, R.; Derolf, Å.; Lehmann, S.; Möllgård, L.; Peterson, S.; Stockelberg, D.; et al. Failure matters: Unsuccessful cytogenetics and unperformed cytogenetics are associated with a poor prognosis in a population-based series of acute myeloid leukaemia. Eur. J. Haematol. 2015, 94, 419–423. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Schroeder, M.C.; O’Laughlin, M.; Wilson, R.; MacMillan, S.; Bohannon, A.; Kruchowski, S.; Garza, J.; Du, F.; Hughes, A.E.; et al. Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. N. Engl. J. Med. 2021, 384, 924–935. [Google Scholar] [CrossRef]
- Sandahl, J.D.; Kjeldsen, E.; Abrahamsson, J.; Ha, S.-Y.; Heldrup, J.; Jahnukainen, K.; Jónsson Ólafur, G.; Lausen, B.; Palle, J.; Zeller, B.; et al. The applicability of the WHO classification in paediatric AML. A NOPHO-AML study. Br. J. Haematol. 2015, 169, 859–867. [Google Scholar] [CrossRef]


| Cytogenetic Subgroups | Fusion Gene or Genes Involved | Frequency in Childhood AML | Median Age (Y) (Range) | Special Features (Age, FAB, Phenotype, Treatment) | Secondary CA | Secondary Molecular Abnormalities | Risk Category | References |
|---|---|---|---|---|---|---|---|---|
| BALANCED CA | ||||||||
| APL | ||||||||
| t(15;17)(q24;q21) | PML-RARA | 6–10% | 12 (1–18) | M3 and M3v, Emergency (DIVC), Specific APL treatment (ATRA, ATO) | tri 8, del(9q), ider(17)(q10) | FLT3-ITD | Favorable | [20,39] |
| CBF leukemias | 20–25% | |||||||
| t(8;21)(q22;q22) | RUNX1-RUNX1T1 | 12–15% | 8 | M2, blasts with single and thin Auer rods, dysgranulopoiesis, CD19+, CD56+ | loss of X or Y, del(9q), tri 8, del(7q), tri 4 | KIT, RAS, FLT3-ITD, FLT3-TKD, ASXL1/2, RAD21 | Favorable | [1,40,41,42,43] |
| inv(16)(p13q22)/ t(16;16)(p13;q22) | CBFB-MYH11 | 5–9% | 9 | M4eo | tri 22, del(7q), tri 8 | KIT, RAS, FLT3-TKD, FLT3-ITD | Favorable | [1,40,41,42,43] |
| 11q23/KMT2Ar | KMT2A with multiple partners | 16–21% | 2.2 (0–18) | M4 and M5, infants | tri 8 | High EVI1 expression, few mutations | Adverse or Intermediate | [44,45,46] |
| t(9;11)(p22;q23) | KMT2A-AF9(MLLT3) | 6–9% | 2.6 | Intermediate | [44,45,46] | |||
| t(11;19)(q23;p13.1) | KMT2A-ELL | 1–2% | 4.6 | Intermediate | [44,45,46] | |||
| t(11;19)(q23;p13.3) | KMT2A-ENL(MLLT1) | 1% | 7.1 | Intermediate | [44,45,46] | |||
| t(10;11)(p12;q23)/ ins(10;11)(p12;q23q13) * | KMT2A-AF10(MLLT10) * | 2–3% | 1.3 | Adverse | [44,45,46] | |||
| t(6;11)(q27;q23) | KMT2A-AF6(MLLT4) | 1–2% | 12.4 | Adverse | [44,45,46] | |||
| 11p15/NUP98r | NUP98 with multiple partners | 3–5% | 11 (1.3–18) | Adverse | [36,47,48] | |||
| t(5;11)(q35;p15) ** | NUP98-NSD1 | 3–4% | 10.4 (1.2–19.4) | M4,M5 71–77% of NUP98r 10–16% of NK | tri 8, del(5q), CK | FLT3-ITD, WT1mut | Adverse | [36,46,47,49,50,51] |
| t(11;12)(p15;p13) ** | NUP98-KMD5A | 1–2% | 3.2 (0.01–18.5) | 10–30% of NUP98r 34% M7, 10% of M7 | CK (numerous numerical and structural CA) | Low frequency of mutations | Adverse | [47,48,52] |
| 12p13 abnormalities | NUP98-KMD5A del(12p) ETV6 (12p13.1) | 4% | Adverse | [22,28] | ||||
| t(7;12)(q36;p13) ** | ETV6; MNX1 | 1% | 0.5 y (0.2–2.3) | Only infants (4% of infants) | tri 19 | unknown | Adverse | [53] |
| Rare other balanced CA | ||||||||
| t(10;11)(p12;q14) | PICALM-MLLT10 | <1% | older children | Extramedullary disease, granulocytic sarcoma, CD7+ | tri 4, tri 19 | Intermediate | [46,50,54] | |
| inv(3)(q21q26.2)/ t(3;3)(q21;q26.2) | GATA2; EVI1(MECOM) | 2% | 3 (2–18) | Dysmyelopoiesis and platelet abnormalities | mon 7 | Adverse | [1,22,24] | |
| t(3;5)(q25;q35) | NPM1-MLF1 | <0.5% | 3.5 (2–13) | M2, M4, M6, dysplasia | rare | unknown | Intermediate | [46,50,55] |
| t(6;9)(p22;q34) | DEK-NUP214 | 1–2% | 12 (2.6–20.4) | M2/M4, dysplasia, basophilia. No infant cases | loss of Y, tri 8, tri 13 | FLT3-ITD | Adverse | [56,57] |
| t(8;16)(p11;p13) | KAT6A-CREBBP | <1% | 1.2 (0–16) | Peak in infants, spontaneous remission in a subset of neonates, DIVC, M4–M5, erythrophagocytosis | tri 1q, del(5q), del(7q), del(9q) | High HOXA9/HOXA10 expression | Intermediate | [50,58] |
| t(16;21)(p11;q22) | FUS-ERG | 0.4% | 8.5 (2.0–17.5) | no | tri 8, tri 10 | Adverse | [50,59] | |
| t(16;21)(q24;q22) | RUNX1-CBFA2T3 | 0.2% | 6.8 (1.0–17) | M1/M2, t-AML | tri 8, loss of Y | Gene expression profile close to RUNX1/RUNX1T1 | Favorable? | [50,59] |
| t(1;22)(p13;q13) | RBM15-MKL1 | 0.3% | 0.7 (0.1–2.7) | Only M7 (5–10% of M7) Hepatosplenomegaly, fibrosis | Mainly no ACA, HD karyotypes | Intermediate | [48,60,61,62,63,64] | |
| inv(16)(p13q24) ** | CBFA2T3-GLIS2 | 2–3% | 1.5 (0.3–17.2) | Infants, 20% of non-DS-AMKL, extramedullary disease, CD56++ | Low HD karyotypes, tri 3, tri 21 | Few mutations | Adverse | [46,48,50,64,65,66,67] |
| t(9;22)(q34;q11) | BCR-ABL1 | 0.6% | Exclude CML-BP or MPAL mBCR Sensitivity to TKI | Association with inv(16)/CBFB-MYH11 | Adverse | [1,14,22] | ||
| UNBALANCED CA | ||||||||
| Monosomy 5, del(5q) | / | 1.2% | 12.5 (0.3–20.7) | M0 | del(17p), CK | Adverse | [7,22,28,68] | |
| Monosomy 7 *** | / | 3% | 7.2 (0–18) | Exclude a primary CA and a predisposition syndrome (GATA2) | / | Adverse | [22,28,69] | |
| del(7q) *** | / | 3% | 7.6 (0–18) | Exclude a primary abnormality and a predisposition syndrome | / | intermediate | [22,28,69] | |
| Trisomy 8 *** | / | 10–14% | 10.1 (0–18) | Mainly a secondary abnormality Search for a primary CA | / | FLT3-ITD | Discussed | [70] |
| Hyperdiploidy (48~49–65 chr.) | tri 8, tri 21, tri 19, tri 6, …. | 11% | 2 (0–17) | AMKL, infants, Search for a primary CA | / | / | No significance | [56,71] |
| Complex karyotype ƒ | / | 8–17% | 3 (0–18) | Exclude a primary CA | / | / | Discussed | [5,6,22,28] |
| Monosomal karyotype ƒƒ | / | 3–5% | 3.6 (0–17) | Exclude a CBF leukemia | / | / | Discussed/ Adverse even after exclusion of mon 7 | [5,6] |
| Normal Karyotype | ||||||||
| Normal karyotype | / | 20–26% | 8.8 (0–18) | Search for a cryptic CA | Search for prognostic mutations: FLT3-ITD, CEBPAdm, NPM1 | According to cryptic CA or to mutations | [7,22,28,36,46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quessada, J.; Cuccuini, W.; Saultier, P.; Loosveld, M.; Harrison, C.J.; Lafage-Pochitaloff, M. Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge. Genes 2021, 12, 924. https://doi.org/10.3390/genes12060924
Quessada J, Cuccuini W, Saultier P, Loosveld M, Harrison CJ, Lafage-Pochitaloff M. Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge. Genes. 2021; 12(6):924. https://doi.org/10.3390/genes12060924
Chicago/Turabian StyleQuessada, Julie, Wendy Cuccuini, Paul Saultier, Marie Loosveld, Christine J. Harrison, and Marina Lafage-Pochitaloff. 2021. "Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge" Genes 12, no. 6: 924. https://doi.org/10.3390/genes12060924
APA StyleQuessada, J., Cuccuini, W., Saultier, P., Loosveld, M., Harrison, C. J., & Lafage-Pochitaloff, M. (2021). Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge. Genes, 12(6), 924. https://doi.org/10.3390/genes12060924