
Formaldehyde and De/Methylation in Age-Related Cognitive Impairment

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Formaldehyde
3. Methylation and Demethylation in DNA, RNA, and Histones
3.1. DNA Methylation
3.2. Histone Modification in Lysine Specific Demethylase (LSD) Releases Two Molecules of Formaldehyde
3.3. RNA Modification
4. Formaldehyde in Epigenetics
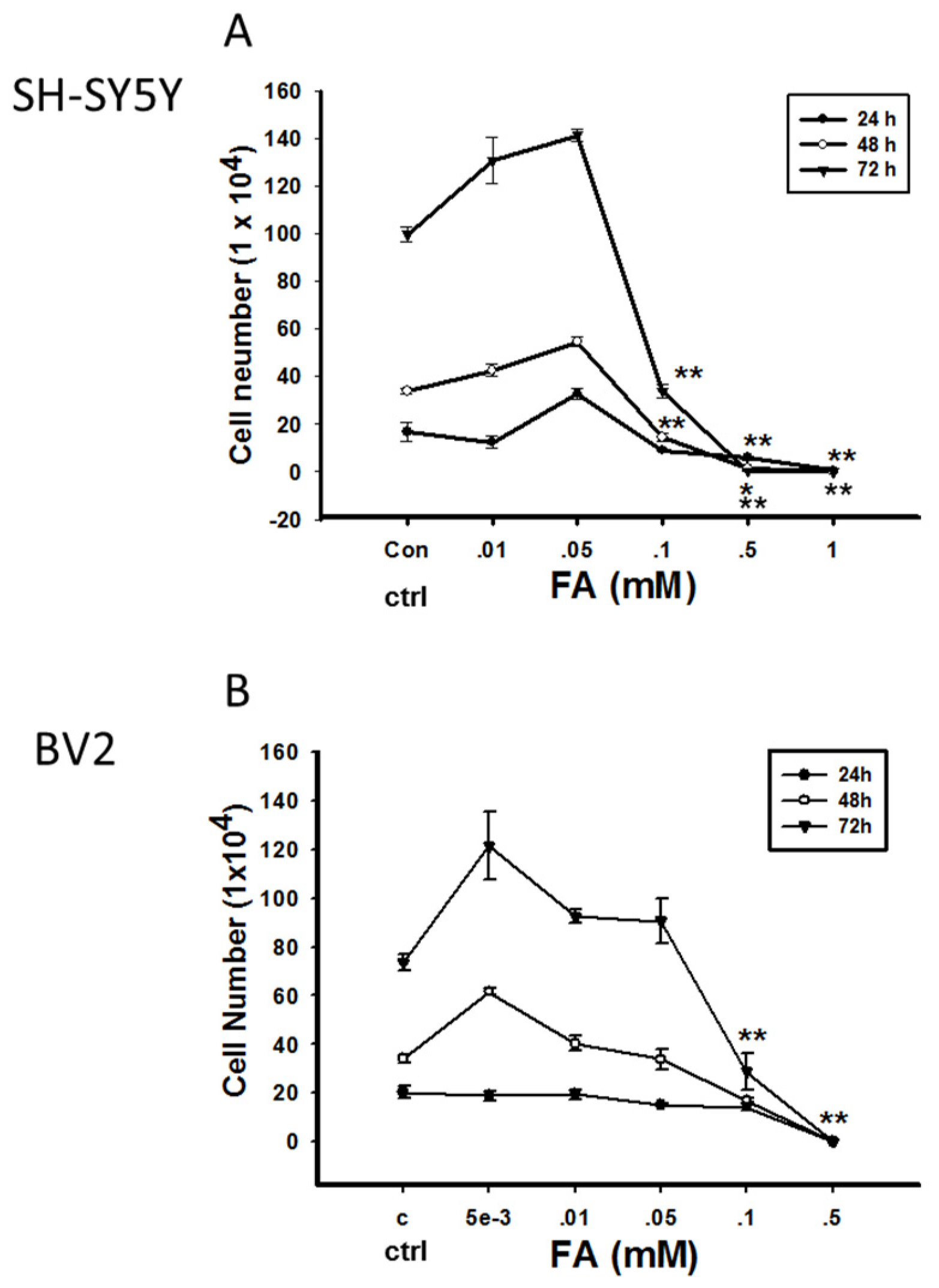
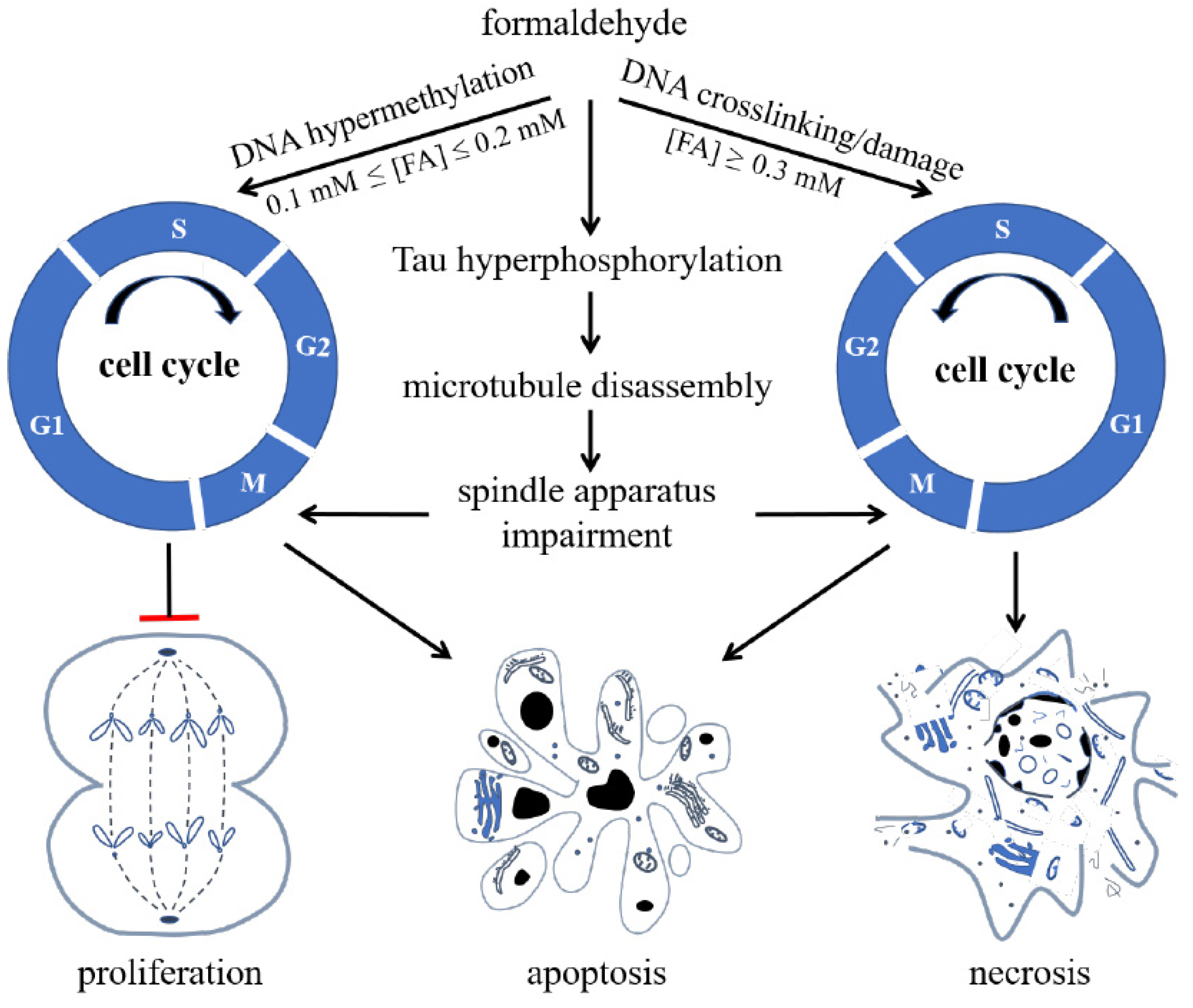
Double-Edged Effects of Formaldehyde
5. Formaldehyde-Involved Epigenetics in Age-Related Cognitive Impairment
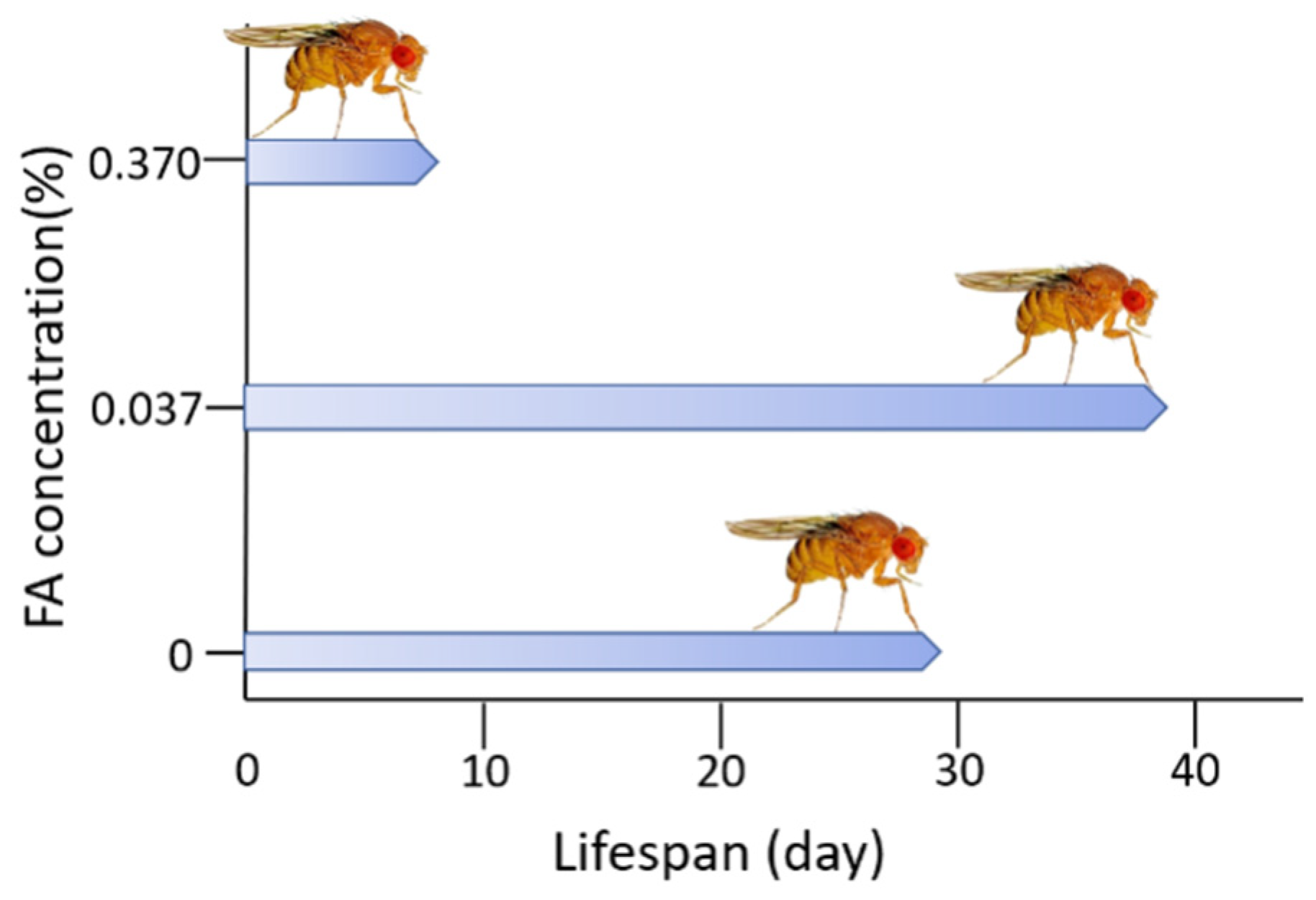
Imbalance of Formaldehyde Metabolism Is Associated with Age-Related Cognitive Impairment
6. Conclusion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FA | formaldehyde |
| ARCI | age-related cognitive impairment |
| AD | Alzheimer’s disease |
| de/methylation | demethylation and methylation |
References
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.Y.; He, R.Q. Effect of acetaldehyde on aggregation of neuronal tau. Protein Pept. Lett. 1999, 6, 105–110. [Google Scholar]
- Yu, P. Involvement of cerebrovascular semicarbazide-sensitive amine oxidase in the pathogenesis of Alzheimer’s disease and vascular dementia. Med. Hypotheses 2001, 57, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; He, R.-Q. Effect of phosphorylation and aggregation on tau binding to DNA. Protein Pept. Lett. 2002, 9, 349–357. [Google Scholar] [CrossRef]
- Nie, C.-L.; Zhang, W.; Zhang, D.; He, R.-Q. Changes in Conformation of Human Neuronal Tau During Denaturation in Formaldehyde Solution. Protein Pept. Lett. 2005, 12, 75–78. [Google Scholar] [CrossRef]
- Nie, C.L.; Wei, Y.; Chen, X.; Liu, Y.Y.; Dui, W.; Liu, Y.; Davies, M.C.; Tendler, S.J.; He, R.G. Formaldehyde at Low Concentration Induces Protein Tau into Globular Amyloid-Like Aggregates In Vitro and In Vivo. PLoS ONE 2007, 2, e629. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Zhang, J.; Luo, W.; Wang, W.; Li, F.; Li, H.; Luo, H.; Lu, J.; Zhou, J.; Wan, Y.; et al. Urine formaldehyde level is inversely correlated to mini mental state examination scores in senile dementia. Neurobiol. Aging 2011, 32, 31–41. [Google Scholar] [CrossRef]
- Lu, J.; Miao, J.-Y.; Pan, R.; He, R.-Q. Formaldehyde-mediated Hyperphosphorylation Disturbs The Interaction Between Tau Protein and DNA*. Prog. Biochem. Biophys. 2011, 38, 1113–1120. [Google Scholar] [CrossRef]
- Lu, J.; Miao, J.; Su, T.; Liu, Y.; He, R. Formaldehyde induces hyperphosphorylation and polymerization of Tau protein both in vitro and in vivo. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 4102–4116. [Google Scholar] [CrossRef]
- He, X.; Li, Z.; Rizak, J.D.; Wu, S.; Wang, Z.; He, R.; Su, M.; Qin, D.; Wang, J.; Hu, X. Resveratrol Attenuates Formaldehyde Induced Hyperphosphorylation of Tau Protein and Cytotoxicity in N2a Cells. Front. Neurosci. 2017, 10, 598. [Google Scholar] [CrossRef]
- Su, T.; Monte, W.C.; Hu, X.; He, Y.; He, R. Formaldehyde as a trigger for protein aggregation and potential target for mitigation of age-related, progressive cognitive impairment. Curr. Top. Med. Chem. 2015, 16, 472–484. [Google Scholar] [CrossRef]
- Axelrod, J. The enzymatic N-demethylation of narcotic drugs. J. Pharmacol. Exp. Ther. 1956, 117, 322–330. [Google Scholar] [PubMed]
- Michalowsky, L.A.; Jones, P.A. DNA Methylation and Differentiation. Environ. Heal. Perspect. 1989, 80, 189. [Google Scholar] [CrossRef]
- Riggs, A. DNA methylation and cell memory. Cell Biophys. 1989, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abeles, R.H.; Frey, P.A.; Jencks, W.P. One Carbon Metabolism. In Biochemistry; Chapter 25, One Carbon Metabolism; Jones and Bartlett Publishers: Boston, MA, USA, 1992. [Google Scholar]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 1999, 70, 288–292. [Google Scholar] [CrossRef]
- Tong, Z.-Q.; Han, C.-S.; Miao, J.-Y.; Lu, J.; He, R.-Q. Excess Endogenous Formaldehyde Induces Memory Decline*. Prog. Biochem. Biophys. 2011, 38, 575–579. [Google Scholar] [CrossRef]
- Tong, Z.; Han, C.; Luo, W.; Li, H.; Luo, H.; Qiang, M.; Su, T.; Wu, B.; Liu, Y.; Yang, X.; et al. Aging-associated excess formaldehyde leads to spatial memory deficits. Sci. Rep. 2013, 3, srep01807. [Google Scholar] [CrossRef]
- Tong, Z.; Han, C.; Qiang, M.; Wang, W.; Lv, J.; Zhang, S.; Luo, W.; Li, H.; Luo, H.; Zhou, J.; et al. Age-related formaldehyde interferes with DNA methyltransferase function, causing memory loss in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 100–110. [Google Scholar] [CrossRef]
- He, R. Cognitive Ability and Impairment Related to Formaldehyde. Formaldehyde Cogn. 2017, 8, 143–166. [Google Scholar]
- Wang, F.; Chen, D.; Wu, P.; Klein, C.; Jin, C. Formaldehyde, Epigenetics, and Alzheimer’s Disease. Chem. Res. Toxicol. 2019, 32, 820–830. [Google Scholar] [CrossRef]
- Bernstein, R.S.; Stayner, L.T.; Elliott, L.J.; Kimbrough, R.; Falk, H.; Blade, L. Inhalation exposure to formaldehyde: An overview of its toxicology, epidemiology, monitoring, and control. Am. Ind. Hyg. Assoc. J. 1984, 45, 778–785. [Google Scholar] [CrossRef]
- Baker, R.R. The generation of formaldehyde in cigarettes—Overview and recent experiments. Food Chem. Toxicol. 2006, 44, 1799–1822. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. P.H.S., National Toxicology Program, Report on Carcinogens, Eleventh ed.; U.S. Department of Health and Human Services: Washington, DC, USA, 2005.
- Shao, Y.; Wang, Y.; Zhao, R.; Chen, J.; Zhang, F.; Linhardt, R.J.; Zhong, W. Biotechnology progress for removal of indoor gaseous formaldehyde. Appl. Microbiol. Biotechnol. 2020, 104, 3715–3727. [Google Scholar] [CrossRef]
- Wartew, G.A. The health hazards of formaldehyde. J. Appl. Toxicol. 1983, 3, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Raja, D.S.; Sultana, B. Potential health hazards for students exposed to formaldehyde in the gross anatomy laboratory. J. Environ. Heal. 2012, 74, 36–40. [Google Scholar]
- Jacobsen, D.; Webb, R.; Collins, T.D.; McMartin, K.E. Methanol and Formate Kinetics in Late Diagnosed Methanol Intoxication. Med. Toxicol. Adverse. Drug Exp. 1988, 3, 418–423. [Google Scholar] [CrossRef]
- Heck, H.D.; Casanova, M. The implausibility of leukemia induction by formaldehyde: A critical review of the biological evidence on distant-site toxicity. Regul. Toxicol. Pharmacol. 2004, 40, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Li, F.X.; Lu, J.; Xu, Y.J.; Tong, Z.Q.; Nie, C.L.; He, R.Q. Formaldehyde-mediated chronic damage may be related to sporadic neurodegeneration. Prog. Biochem. Biophys. 2008, 35, 393–400. [Google Scholar]
- Walport, L.J.; Hopkinson, R.J.; Schofield, C.J. Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 2012, 16, 525–534. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, C.; Chen, Y.; Yu, L.; Li, Y.; Wei, Y.; Li, W.; He, R. Formaldehyde produced from d-ribose under neutral and alkaline conditions. Toxicol. Rep. 2019, 6, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Gleisten, M.P.; Donohue, T.J. Identification of proteins involved in formaldehyde metabolism by Rhodobacter sphaeroides. Microbiology 2008, 154, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Dicker, E.; Cederbaum, A.I. Inhibition of CO2 production from aminopyrine or methanol by cyanamide or crotonaldehyde and the role of mitochondrial aldehyde dehydrogenase in formaldehyde oxidation. Biochim. Biophys. Acta (BBA) Gen. Subj. 1986, 883, 91–97. [Google Scholar] [CrossRef]
- Koivusalo, M.; Baumann, M.; Uotila, L. Evidence for the identity of glutathione-dependent formaldehyde dehydrogenase and class III alcohol dehydrogenase. FEBS Lett. 1989, 257, 105–109. [Google Scholar] [CrossRef]
- MacAllister, S.L.; Choi, J.; Dedina, L.; O’Brien, P.J. Metabolic mechanisms of methanol/formaldehyde in isolated rat hepatocytes: Carbonyl-metabolizing enzymes versus oxidative stress. Chem. Inter. 2011, 191, 308–314. [Google Scholar] [CrossRef]
- Dorokhov, Y.L.; Shindyapina, A.; Sheshukova, E.V.; Komarova, T.V. Metabolic Methanol: Molecular Pathways and Physiological Roles. Physiol. Rev. 2015, 95, 603–644. [Google Scholar] [CrossRef]
- Burgos-Barragan, G.; Wit, N.; Meiser, J.; Dingler, F.A.; Pietzke, M.; Mulderrig, L.; Pontel, L.B.; Rosado, I.V.; Brewer, T.F.; Cordell, R.L.; et al. Erratum: Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nat. Cell Biol. 2017, 548, 612. [Google Scholar] [CrossRef]
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone Methylation Regulates Memory Formation. J. Neurosci. 2010, 30, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Dezi, V.; Ivanov, C.; Haussmann, I.U.; Soller, M. Nucleotide modifications in messenger RNA and their role in development and disease. Biochem. Soc. Trans. 2016, 44, 1385–1393. [Google Scholar] [CrossRef]
- Bochtler, M.; Kolano, A.; Xu, G.-L. DNA demethylation pathways: Additional players and regulators. BioEssays 2017, 39, e201600178-13. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, Mechanisms and Clinical Perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Allis, C.D.; Caparros, M.L.; Jenuwein, T.; Reinberg, D. Epigenetics; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015. [Google Scholar]
- Barros, S.; Offenbacher, S. Epigenetics: Connecting Environment and Genotype to Phenotype and Disease. J. Dent. Res. 2009, 88, 400–408. [Google Scholar] [CrossRef]
- Razin, A.; Riggs, A. DNA methylation and gene function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA Methylation in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Pinney, S.E. Mammalian Non-CpG Methylation: Stem Cells and Beyond. Biology 2014, 3, 739–751. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- van der Wijst, M.G.; Venkiteswaran, M.; Chen, H.; Xu, G.L.; Plösch, T.; Rots, M.G. Local chromatin microenvironment determines DNMT activity: From DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics 2015, 10, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-Aza-Deoxycytidine Induces Selective Degradation of DNA Methyltransferase 1 by a Proteasomal Pathway That Requires the KEN Box, Bromo-Adjacent Homology Domain, and Nuclear Localization Signal. Mol. Cell. Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Tahiliani, M. Expanding the Epigenetic Landscape: Novel Modifications of Cytosine in Genomic DNA. Cold Spring Harb. Perspect. Biol. 2014, 6, a018630. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; Hong, C.; Xing, X.; Zhou, X.; Li, D.; Coarfa, C.; Bell, R.J.; Maire, C.L.; Ligon, K.L.; Sigaroudinia, M.; et al. Intermediate DNA methylation is a conserved signature of genome regulation. Nat. Commun. 2015, 6, 6363. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B.; Bates, P.A.; Paik, J.; Jacobs, S.C.; Lindahl, T. Repair of alkylated DNA: Recent advances. DNA Repair 2007, 6, 429–442. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Beaudet, A.L. Epigenetics and Human Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a019497. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nat. Cell Biol. 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. SnapShot: Histone Modifications. Cell 2014, 159, 458–458.e1. [Google Scholar] [CrossRef]
- Smith, B.C.; Denu, J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2009, 1789, 45–57. [Google Scholar] [CrossRef]
- Musselman, C.A.; LaLonde, M.-E.; Côté, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fu, J.; Zhou, Y. A Review in Research Progress Concerning m6A Methylation and Immunoregulation. Front. Immunol. 2019, 10, 922. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. Erratum: Corrigendum: N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2012, 8, 1008. [Google Scholar] [CrossRef]
- Sánchez-Pulido, L.; Andrade-Navarro, M.A. The FTO (fat mass and obesity associated) gene codes for a novel member of the non-heme dioxygenase superfamily. BMC Biochem. 2007, 8, 23. [Google Scholar] [CrossRef]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.B.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A Common Variant in the FTO Gene Is Associated with Body Mass Index and Predisposes to Childhood and Adult Obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Gerken, T.; Girard, C.A.; Tung, Y.-C.L.; Webby, C.J.; Saudek, V.; Hewitson, K.S.; Yeo, G.S.H.; McDonough, M.A.; Cunliffe, S.; McNeill, L.A.; et al. The Obesity-Associated FTO Gene Encodes a 2-Oxoglutarate-Dependent Nucleic Acid Demethylase. Science 2007, 318, 1469–1472. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, B.F.; Sun, Y.; Shi, X.; Yang, W.; Xiao, Y.J.; Hao, X.L.; Ping, Y.S.; Chen, W.J.; Wang, K.X.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Toh, J.D.W.; Crossley, S.W.M.; Bruemmer, K.J.; Ge, E.J.; He, D.; Iovan, D.A.; Chang, C.J. Distinct RNA N-demethylation pathways catalyzed by nonheme iron ALKBH5 and FTO enzymes enable regulation of formaldehyde release rates. Proc. Natl. Acad. Sci. USA 2020, 117, 25284–25292. [Google Scholar] [CrossRef]
- Zhou, Z.; Lv, J.; Yu, H.; Han, J.; Yang, X.; Feng, D.; Wu, Q.; Yuan, B.; Lu, Q.; Yang, H. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol. Cancer 2020, 19, 1–20. [Google Scholar] [CrossRef]
- Esteve-Puig, R.; Bueno-Costa, A.; Esteller, M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett. 2020, 474, 127–137. [Google Scholar] [CrossRef]
- Costa, F.F. Non-coding RNAs, epigenetics and complexity. Gene 2008, 410, 9–17. [Google Scholar] [CrossRef]
- Wei, J.-W.; Huang, K.; Yang, C.; Kang, C.-S. Non-coding RNAs as regulators in epigenetics. Oncol. Rep. 2016, 37, 3–99. [Google Scholar] [CrossRef]
- Francia, S. Non-Coding RNA: Sequence-Specific Guide for Chromatin Modification and DNA Damage Signaling. Front. Genet. 2015, 6, 320. [Google Scholar] [CrossRef] [PubMed]
- Morellato, A.E.; Umansky, C.; Pontel, L.B. The toxic side of one-carbon metabolism and epigenetics. Redox Biol. 2021, 40, 101850. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.H.; Lu, L.-X.; Fan, H.; Kazachkov, M.; Jiang, Z.-J.; Jalkanen, S.; Stolen, C. Involvement of Semicarbazide-Sensitive Amine Oxidase-Mediated Deamination in Lipopolysaccharide-Induced Pulmonary Inflammation. Am. J. Pathol. 2006, 168, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yang, L.; Gong, C.; Tao, G.; Huang, H.; Liu, J.; Zhang, H.; Wu, D.; Xia, B.; Hu, G.; et al. Effects of long-term low-dose formaldehyde exposure on global genomic hypomethylation in 16HBE cells. Toxicol. Lett. 2011, 205, 235–240. [Google Scholar] [CrossRef]
- Yoshida, I.; Ibuki, Y. Formaldehyde-induced histone H3 phosphorylation via JNK and the expression of proto-oncogenes. Mutat. Res. Mol. Mech. Mutagen. 2014, 770, 9–18. [Google Scholar] [CrossRef]
- Simonsson, M.; Heldin, C.-H.; Ericsson, J.; Gronroos, E. The Balance between Acetylation and Deacetylation Controls Smad7 Stability. J. Biol. Chem. 2005, 280, 21797–21803. [Google Scholar] [CrossRef]
- Rager, J.E.; Moeller, B.C.; Miller, S.K.; Kracko, D.; Doyle-Eisele, M.; Swenberg, J.A.; Fry, R.C. Formaldehyde-Associated Changes in microRNAs: Tissue and Temporal Specificity in the Rat Nose, White Blood Cells, and Bone Marrow. Toxicol. Sci. 2013, 138, 36–46. [Google Scholar] [CrossRef]
- Li, G.; Yang, J.; Ling, S. Formaldehyde exposure alters miRNA expression profiles in the olfactory bulb. Inhal. Toxicol. 2015, 27, 387–393. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.; Mo, W.C.; He, Y.G.; Wei, Y.; He, R.Q.; Yi, F.P. Accumulation of Simulated Pathological Level of Formaldehyde Decreases Cell Viability and Adhesive Morphology in Neuronal Cells. Prog. Biochem. Biophys. 2017, 44, 601–614. [Google Scholar]
- Hou, Q.; Jiang, H.; Zhang, X.; Guo, C.; Huang, B.; Wang, P.; Wang, T.; Wu, K.; Li, J.; Gong, Z.; et al. Nitric oxide metabolism controlled by formaldehyde dehydrogenase (fdh, homolog of mammalian GSNOR) plays a crucial role in visual pattern memory in Drosophila. Nitric Oxide 2011, 24, 17–24. [Google Scholar] [CrossRef]
- Wu, K.; Ren, R.; Su, W.; Wen, B.; Zhang, Y.; Yi, F.; Qiao, X.; Yuan, T.; Wang, J.; Liu, L.; et al. A Novel Suppressive Effect of Alcohol Dehydrogenase 5 in Neuronal Differentiation. J. Biol. Chem. 2014, 289, 20193–20199. [Google Scholar] [CrossRef]
- Li, Y.N.; He, R.Q. The Effects of Formaldehyde on Life Span and Stress Resistance in Drosophila melanogaster. Prog. Biochem. Biophys. 2016, 43, 420–428. [Google Scholar]
- Scheltens, P.; Strooper, B.D.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar]
- Sanabria-Castro, A.; Alvarado-Echeverria, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Lill, C.; Tanzi, R.E. The Genetics of Alzheimer Disease: Back to the Future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Bohnuud, T.; Beglov, D.; Ngan, C.H.; Zerbe, B.; Hall, D.R.; Brenke, R.; Vajda, S.; Frank-Kamenetskii, M.D.; Kozakov, D. Computational mapping reveals dramatic effect of Hoogsteen breathing on duplex DNA reactivity with formaldehyde. Nucleic Acids Res. 2012, 40, 7644–7652. [Google Scholar] [CrossRef]
- Lu, K.; Craft, S.; Nakamura, J.; Moeller, B.C.; Swenberg, J.A. Use of LC-MS/MS and stable isotopes to differentiate hydroxymethyl and methyl DNA adducts from formaldehyde and nitrosodimethylamine. Chem. Res. Toxicol. 2012, 25, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.C. Role of DNA Methylation in the Regulation of Cell Function: Autoimmunity, Aging and Cancer. J. Nutr. 2002, 132, 2401S–2405S. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Miao, J.-Y.; Lu, J.; Zhang, Z.-J.; Tong, Z.-Q.; He, R.-Q. The Effect of Formaldehyde on Cell Cycle Is in a Concentration-dependent Manner. Acta Agron. Sin. 2013, 40, 641. [Google Scholar] [CrossRef]
- Pei, Y.; Davies, J.; Zhang, M.; Zhang, H.-T. The Role of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 76, 49–62. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Kalasz, H. Biological Role of Formaldehyde, and Cycles Related to Methylation, Demethylation, and Formaldehyde Production. Mini Revi. Med. Chem. 2003, 3, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.F.; Lu, J.; Miao, J.Y.; Rizak, J.; Yang, J.Z.; Zhai, R.W.; Zhou, J.; Qu, J.G.; Wang, J.H.; Yang, S.C.; et al. Alzheimer’s Disease and Methanol Toxicity (Part 1): Chronic Methanol Feeding Led to Memory Impairments and Tau Hyperphosphorylation in Mice. J. Alzheimers Dis. 2014, 41, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Miao, J.; Rizak, R.; Zhai, Z.; Wang, T.; Huma, T.; Li, N.; Zheng, S.; Wu, Y.; Zheng, X.; et al. Alzheimer’s disease and methanol toxicity (part 2): Lessons from four rhesus macaques (Macaca mulatta) chronically fed methanol. J. Alzheimers Dis. 2014, 41, 1131–1147. [Google Scholar] [CrossRef]
- Lee, J.C.; Kim, S.J.; Hong, S.; Kim, Y. Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Li, Z.H.; He, X.P.; Li, H.; He, R.Q.; Hu, X.T. Age-associated changes in amyloid-beta and formaldehyde concentrations in cerebrospinal fluid of rhesus monkeys. Zool. Res. 2020, 41, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Zhang, Y.; Mei, X.; Yue, W.; Jiang, L.; Ai, Y.; Yu, H.; Luo, H.; Li, W.; Luo, X.; et al. Degradation of FA reduces Abeta neurotoxicity and Alzheimer-related phenotypes. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Tong, Z.; Han, C.; Luo, W.; Wang, X.; Li, H.; Luo, H.; Zhou, J.; Qi, J.; He, R. Accumulated hippocampal formaldehyde induces age-dependent memory decline. AGE 2012, 35, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Wu, R.; Ye, M.; Zhao, Y.; Kang, J.; Ma, P.; Li, J.; Yang, X. Acute formaldehyde exposure induced early Alzheimer-like changes in mouse brain. Toxicol. Mech. Methods 2017, 28, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Tagliatesta, S.; Caiafa, P.; Zampieri, M. DNA methylation dynamics in aging: How far are we from understanding the mechanisms? Mech. Ageing Dev. 2018, 174, 3–17. [Google Scholar] [CrossRef]
- Wilkinson, G.S.; Adams, D.M.; Haghani, A.; Lu, A.T.; Zoller, J.; Breeze, C.E.; Arnold, B.D.; Ball, H.C.; Carter, G.G.; Cooper, D.K.N.; et al. DNA methylation predicts age and provides insight into exceptional longevity of bats. Nat. Commun. 2021, 12, 1615. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V.W. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef]
- Sanchez-Mut, J.V.; Graff, J. Epigenetic Alterations in Alzheimer’s Disease. Front. Behav. Neurosci. 2015, 9, 347. [Google Scholar] [CrossRef]
- Zhang, Z. Long non-coding RNAs in Alzheimer’s disease. Curr. Top Med. Chem. 2016, 16, 511–519. [Google Scholar] [CrossRef]
- Dehghani, R.; Rahmani, F.; Rezaei, N. MicroRNA in Alzheimer’s disease revisited: Implications for major neuropathological mechanisms. Rev. Neurosci. 2017, 29, 161–182. [Google Scholar] [CrossRef]
- Xiao, R.; He, R.Q. Metabolism of Formaldehyde In Vivo. Formaldehyde and Cognition, 1st ed.; Chapter 8; Springer: Amsterdam, The Netherlands, 2017; pp. 21–46. [Google Scholar]
- Liu, K.L.; He, Y.G.; Yu, L.X.; He, R.Q. Elevated formaldehyde in the cecum of APP/PS1 mouse. Microbiol. China 2017, 44, 1761–1766. [Google Scholar]
- Mou, L.X.; Hu, P.D.; Cao, X.; He, R.Q. Changes in intestinal lengths of 129S2/SvPasCrl mice from adulthood to agedness. Microbiol. China 2021, 48, 1057–1060. [Google Scholar]
- Kennedy, D.O. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.F.; Jiang, C.; Wan, Y.; Lv, J.H.; Jia, J.P.; Wang, X.M.; Yang, X.; Tong, Z.Q. Aging-associated formaldehyde-induced norepinephrine deficiency contributes to age-related memory decline. Aging Cell 2015, 14, 659–668. [Google Scholar] [CrossRef]
- Qu, M.H.; He, R.Q. Formaldehyde from Environment. Formaldehyde and Cognition, 1st ed.; Chapter 8; Springer: Amsterdam, The Netherlands; pp. 1–20.



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Wei, Y.; Qu, M.; Mou, L.; Miao, J.; Xi, M.; Liu, Y.; He, R. Formaldehyde and De/Methylation in Age-Related Cognitive Impairment. Genes 2021, 12, 913. https://doi.org/10.3390/genes12060913
Li T, Wei Y, Qu M, Mou L, Miao J, Xi M, Liu Y, He R. Formaldehyde and De/Methylation in Age-Related Cognitive Impairment. Genes. 2021; 12(6):913. https://doi.org/10.3390/genes12060913
Chicago/Turabian StyleLi, Ting, Yan Wei, Meihua Qu, Lixian Mou, Junye Miao, Mengqi Xi, Ying Liu, and Rongqiao He. 2021. "Formaldehyde and De/Methylation in Age-Related Cognitive Impairment" Genes 12, no. 6: 913. https://doi.org/10.3390/genes12060913
APA StyleLi, T., Wei, Y., Qu, M., Mou, L., Miao, J., Xi, M., Liu, Y., & He, R. (2021). Formaldehyde and De/Methylation in Age-Related Cognitive Impairment. Genes, 12(6), 913. https://doi.org/10.3390/genes12060913