
Challenges for Cryptosporidium Population Studies


Abstract
1. Introduction
2. Current Status of Cryptosporidium Whole Genome Sequences
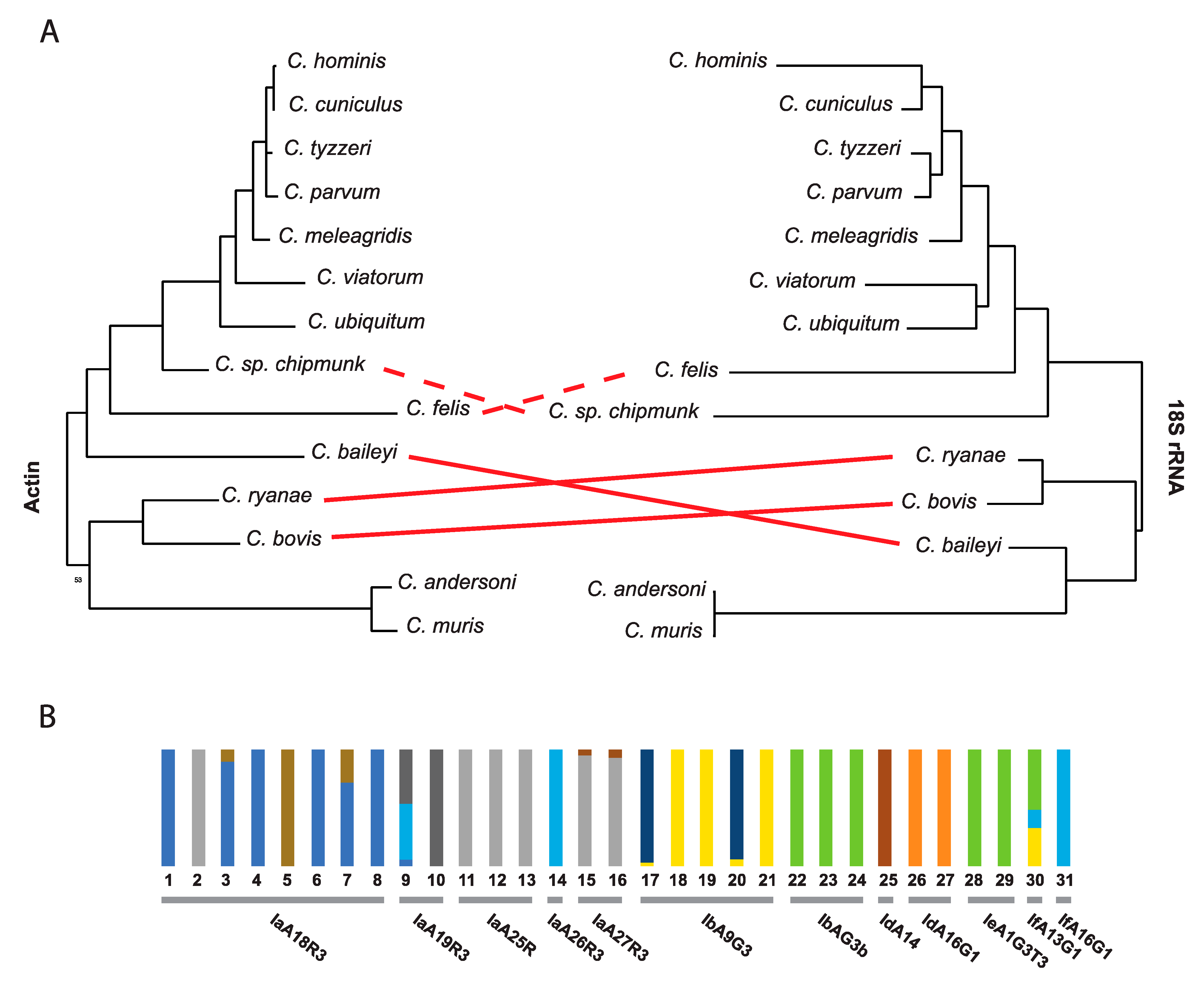
3. Cryptosporidium Population Structure
4. Challenges Faced by Cryptosporidium Population Studies
4.1. Sampling Limitations
4.2. Limited Number of Markers
4.3. Mixed Infections
4.4. Detection of Sexual Recombination in Cryptosporidium
4.5. Lack of Metadata for Global Comparative Studies
5. Detecting Mixed Populations in Collected Samples
6. Emerging Solutions to Deal with This Challenging Parasite
6.1. Promising In Vitro Cultivation Systems Parasites
6.2. Sorted Single-Cell Genomic Sequencing
6.3. Cryptosporidium Capture Enrichment Sequencing
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- Khalil, I.A.; Troeger, C.; Rao, P.C.; Blacker, B.F.; Brown, A.; Brewer, T.G.; Colombara, D.V.; De Hostos, E.L.; Engmann, C.; Guerrant, R.L.; et al. Morbidity, mortality, and long-term consequences associated with diarrhoea from Cryptosporidium infection in children younger than 5 years: A meta-analyses study. Lancet Glob. Health 2018, 6, e758–e768. [Google Scholar] [CrossRef]
- Feng, Y.; Ryan, U.M.; Xiao, L. Genetic Diversity and Population Structure of Cryptosporidium. Trends Parasitol. 2018, 34, 997–1011. [Google Scholar] [CrossRef]
- Xiao, L.; Feng, Y. Molecular epidemiologic tools for waterborne pathogens Cryptosporidium spp. and Giardia duodenalis. Food Waterborne Parasitol. 2017, 8, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Current, W.L.; Reese, N.C. A comparison of endogenous development of three isolates of Cryptosporidium in suckling mice. J. Protozool. 1986, 33, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Tandel, J.; English, E.D.; Sateriale, A.; Gullicksrud, J.A.; Beiting, D.P.; Sullivan, M.C.; Pinkston, B.; Striepen, B. Life cycle progression and sexual development of the apicomplexan parasite Cryptosporidium parvum. Nat. Microbiol. 2019, 4, 2226–2236. [Google Scholar] [CrossRef]
- Cama, V.; Gilman, R.H.; Vivar, A.; Ticona, E.; Ortega, Y.; Bern, C.; Xiao, L. Mixed Cryptosporidium infections and HIV. Emerg. Infect. Dis. 2006, 12, 1025–1028. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Cotton, J.A.; Burkey, C.; Arju, T.; Gilmartin, A.; Lin, Y.; Ahmed, E.; Steiner, K.; Alam, M.; Ahmed, S.; et al. Genetic diversity of Cryptosporidium hominis in a Bangladeshi community as revealed by whole genome sequencing. J. Infect. Dis. 2018, 218, 259–264. [Google Scholar] [CrossRef]
- Korpe, P.S.; Gilchrist, C.; Burkey, C.; Taniuchi, M.; Ahmed, E.; Madan, V.; Castillo, R.; Ahmed, S.; Arju, T.; Alam, M.; et al. Case-Control Study of Cryptosporidium Transmission in Bangladeshi Households. Clin. Infect. Dis. 2019, 68, 1073–1079. [Google Scholar] [CrossRef]
- Sannella, A.R.; Suputtamongkol, Y.; Wongsawat, E.; Caccio, S.M. A retrospective molecular study of Cryptosporidium species and genotypes in HIV-infected patients from Thailand. Parasites Vectors 2019, 12, 91. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, S.; Zhao, W.; Guo, Y.; Li, N.; Zheng, Z.; Zhang, L.; Kvac, M.; Xiao, L.; Feng, Y. Population structure and geographical segregation of Cryptosporidium parvum IId subtypes in cattle in China. Parasites Vectors 2020, 13, 425. [Google Scholar] [CrossRef] [PubMed]
- Ramo, A.; Quilez, J.; Monteagudo, L.; Del Cacho, E.; Sanchez-Acedo, C. Intra-Species Diversity and Panmictic Structure of Cryptosporidium parvum Populations in Cattle Farms in Northern Spain. PLoS ONE 2016, 11, e0148811. [Google Scholar] [CrossRef]
- Ramo, A.; Monteagudo, L.V.; Del Cacho, E.; Sanchez-Acedo, C.; Quilez, J. Intra-Species Genetic Diversity and Clonal Structure of Cryptosporidium parvum in Sheep Farms in a Confined Geographical Area in Northeastern Spain. PLoS ONE 2016, 11, e0155336. [Google Scholar] [CrossRef]
- Morrison, L.J.; Mallon, M.E.; Smith, H.V.; MacLeod, A.; Xiao, L.; Tait, A. The population structure of the Cryptosporidium parvum population in Scotland: A complex picture. Infect. Genet. Evol. 2008, 8, 121–129. [Google Scholar] [CrossRef]
- Widmer, G.; Carmena, D.; Kvac, M.; Chalmers, R.M.; Kissinger, J.C.; Xiao, L.; Sateriale, A.; Striepen, B.; Laurent, F.; Lacroix-Lamande, S.; et al. Update on Cryptosporidium spp.: Highlights from the Seventh International Giardia and Cryptosporidium Conference. Parasite 2020, 27, 14. [Google Scholar] [CrossRef]
- Strong, W.B.; Gut, J.; Nelson, R.G. Cloning and sequence analysis of a highly polymorphic Cryptosporidium parvum gene encoding a 60-kilodalton glycoprotein and characterization of its 15- and 45-kilodalton zoite surface antigen products. Infect. Immun. 2000, 68, 4117–4134. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.; Putignani, L.; McLauchlin, J.; Casemore, D.P.; Crisanti, A. PCR-RFLP analysis of the Cryptosporidium oocyst wall protein (COWP) gene discriminates between C. wrairi and C. parvum, and between C. parvum isolates of human and animal origin. FEMS Microbiol. Lett. 1997, 150, 209–217. [Google Scholar] [CrossRef]
- Xiao, L.; Morgan, U.M.; Limor, J.; Escalante, A.; Arrowood, M.; Shulaw, W.; Thompson, R.C.; Fayer, R.; Lal, A.A. Genetic diversity within Cryptosporidium parvum and related Cryptosporidium species. Appl. Environ. Microbiol. 1999, 65, 3386–3391. [Google Scholar] [CrossRef]
- Robinson, G.; Chalmers, R.M. Assessment of polymorphic genetic markers for multi-locus typing of Cryptosporidium parvum and Cryptosporidium hominis. Exp. Parasitol. 2012, 132, 200–215. [Google Scholar] [CrossRef]
- Li, N.; Xiao, L.; Cama, V.A.; Ortega, Y.; Gilman, R.H.; Guo, M.; Feng, Y. Genetic recombination and Cryptosporidium hominis virulent subtype IbA10G2. Emerg. Infect. Dis. 2013, 19, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.; Robinson, G.; Swain, M.T.; Chalmers, R.M. Direct Sequencing of Cryptosporidium in Stool Samples for Public Health. Front. Public Health 2019, 7, 360. [Google Scholar] [CrossRef] [PubMed]
- Nader, J.L.; Mathers, T.C.; Ward, B.J.; Pachebat, J.A.; Swain, M.T.; Robinson, G.; Chalmers, R.M.; Hunter, P.R.; Oosterhout, C.; Tyler, K.M. Evolutionary genomics of anthroponosis in Cryptosporidium. Nat. Microbiol. 2019, 4, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Tichkule, S.; Jex, A.R.; van Oosterhout, C.; Sannella, A.R.; Krumkamp, R.; Aldrich, C.; Maiga-Ascofare, O.; Dekker, D.; Lamshoft, M.; Mbwana, J.; et al. Comparative genomics revealed adaptive admixture in Cryptosporidium hominis in Africa. Microb. Genom. 2021, 7, mgen000493. [Google Scholar] [CrossRef]
- Kissinger, J.C.; DeBarry, J. Genome cartography: Charting the apicomplexan genome. Trends Parasitol. 2011, 27, 345–354. [Google Scholar] [CrossRef]
- Xu, P.; Widmer, G.; Wang, Y.; Ozaki, L.S.; Alves, J.M.; Serrano, M.G.; Puiu, D.; Manque, P.; Akiyoshi, D.; Mackey, A.J.; et al. The genome of Cryptosporidium hominis. Nature 2004, 431, 1107–1112. [Google Scholar] [CrossRef]
- Abrahamsen, M.S.; Templeton, T.J.; Enomoto, S.; Abrahante, J.E.; Zhu, G.; Lancto, C.A.; Deng, M.; Liu, C.; Widmer, G.; Tzipori, S.; et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science 2004, 304, 441–445. [Google Scholar] [CrossRef]
- Piper, M.B.; Bankier, A.T.; Dear, P.H. A HAPPY map of Cryptosporidium parvum. Genome Res. 1998, 8, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Seeber, F.; Limenitakis, J.; Soldati-Favre, D. Apicomplexan mitochondrial metabolism: A story of gains, losses and retentions. Trends Parasitol. 2008, 24, 468–478. [Google Scholar] [CrossRef]
- Zhu, G.; Marchewka, M.J.; Keithly, J.S. Cryptosporidium parvum appears to lack a plastid genome. Microbiology 2000, 146, 315–321. [Google Scholar] [CrossRef]
- Ifeonu, O.O.; Chibucos, M.C.; Orvis, J.; Su, Q.; Elwin, K.; Guo, F.; Zhang, H.; Xiao, L.; Sun, M.; Chalmers, R.M.; et al. Annotated draft genome sequences of three species of Cryptosporidium: Cryptosporidium meleagridis isolate UKMEL1, C. baileyi isolate TAMU-09Q1 and C. hominis isolates TU502_2012 and UKH1. Pathog. Dis. 2016, 74, ftw080. [Google Scholar] [CrossRef]
- Sateriale, A.; Slapeta, J.; Baptista, R.; Engiles, J.B.; Gullicksrud, J.A.; Herbert, G.T.; Brooks, C.F.; Kugler, E.M.; Kissinger, J.C.; Hunter, C.A.; et al. A Genetically Tractable, Natural Mouse Model of Cryptosporidiosis Offers Insights into Host Protective Immunity. Cell Host Microbe 2019, 26, 135–146.e5. [Google Scholar] [CrossRef]
- Xu, Z.; Guo, Y.; Roellig, D.M.; Feng, Y.; Xiao, L. Comparative analysis reveals conservation in genome organization among intestinal Cryptosporidium species and sequence divergence in potential secreted pathogenesis determinants among major human-infecting species. BMC Genom. 2019, 20, 406. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Roellig, D.M.; Guo, Y.; Li, N.; Frace, M.A.; Tang, K.; Zhang, L.; Feng, Y.; Xiao, L. Evolution of mitosome metabolism and invasion-related proteins in Cryptosporidium. BMC Genom. 2016, 17, 1006. [Google Scholar] [CrossRef] [PubMed]
- Baptista, R.P.; Li, Y.; Sateriale, A.; Sanders, M.J.; Brooks, K.L.; Tracey, A.; Ansell, B.R.E.; Jex, A.R.; Cooper, G.W.; Smith, E.D.; et al. Long-read assembly and comparative evidence-based reanalysis of Cryptosporidium genome sequences reveal new biological insights. bioRxiv 2021. [Google Scholar] [CrossRef]
- Isaza, J.P.; Galvan, A.L.; Polanco, V.; Huang, B.; Matveyev, A.V.; Serrano, M.G.; Manque, P.; Buck, G.A.; Alzate, J.F. Revisiting the reference genomes of human pathogenic Cryptosporidium species: Reannotation of C. parvum Iowa and a new C. hominis reference. Sci. Rep. 2015, 5, 16324. [Google Scholar] [CrossRef]
- Li, Y.; Baptista, R.P.; Sateriale, A.; Striepen, B.; Kissinger, J.C. Analysis of Long Non-Coding RNA in Cryptosporidium parvum Reveals Significant Stage-Specific Antisense Transcription. Front Cell Infect. Microbiol. 2020, 10, 608298. [Google Scholar] [CrossRef]
- Peng, M.M.; Xiao, L.; Freeman, A.R.; Arrowood, M.J.; Escalante, A.A.; Weltman, A.C.; Ong, C.S.; Mac Kenzie, W.R.; Lal, A.A.; Beard, C.B. Genetic polymorphism among Cryptosporidium parvum isolates: Evidence of two distinct human transmission cycles. Emerg. Infect. Dis. 1997, 3, 567–573. [Google Scholar] [CrossRef]
- Sulaiman, I.I.M.; Lal, A.A.A.; Xiao, L.L. A population genetic study of the Cryptosporidium parvum human genotype parasites. J. Eukaryot. Microbiol. 2001, 48, 24s–27s. [Google Scholar] [CrossRef]
- Sulaiman, I.M.; Lal, A.A.; Xiao, L. Molecular phylogeny and evolutionary relationships of Cryptosporidium parasites at the actin locus. J. Parasitol. 2002, 88, 388–394. [Google Scholar] [CrossRef]
- Xiao, L.; Singh, A.; Limor, J.; Graczyk, T.K.; Gradus, S.; Lal, A. Molecular characterization of Cryptosporidium oocysts in samples of raw surface water and wastewater. Appl. Environ. Microbiol. 2001, 67, 1097–1101. [Google Scholar] [CrossRef]
- Cacciò, S.; Homan, W.; Camilli, R.; Traldi, G.; Kortbeek, T.; Pozio, E. A microsatellite marker reveals population heterogeneity within human and animal genotypes of Cryptosporidium parvum. Parasitology 2000, 120, 237–244. [Google Scholar] [CrossRef]
- Caccio, S.; Spano, F.; Pozio, E. Large sequence variation at two microsatellite loci among zoonotic (genotype C) isolates of Cryptosporidium parvum. Int. J. Parasitol. 2001, 31, 1082–1086. [Google Scholar] [CrossRef]
- Mallon, M.; MacLeod, A.; Wastling, J.; Smith, H.; Reilly, B.; Tait, A. Population structures and the role of genetic exchange in the zoonotic pathogen Cryptosporidium parvum. J. Mol. Evol. 2003, 56, 407–417. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, W.; Ryan, U.; Zhang, L.; Kvac, M.; Koudela, B.; Modry, D.; Li, N.; Fayer, R.; Xiao, L. Development of a multilocus sequence tool for typing Cryptosporidium muris and Cryptosporidium andersoni. J. Clin. Microbiol. 2011, 49, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, R.M.; Robinson, G.; Hotchkiss, E.; Alexander, C.; May, S.; Gilray, J.; Connelly, L.; Hadfield, S.J. Suitability of loci for multiple-locus variable-number of tandem-repeats analysis of Cryptosporidium parvum for inter-laboratory surveillance and outbreak investigations. Parasitology 2016, 144, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cordon, G.; Robinson, G.; Nader, J.; Chalmers, R.M. Discovery of new variable number tandem repeat loci in multiple Cryptosporidium parvum genomes for the surveillance and investigation of outbreaks of cryptosporidiosis. Exp. Parasitol. 2016, 169, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, L.; Axen, C.; Bjorkman, C.; Jian, F.; Amer, S.; Liu, A.; Feng, Y.; Li, G.; Lv, C.; et al. Cryptosporidium parvum IId family: Clonal population and dispersal from Western Asia to other geographical regions. Sci. Rep. 2014, 4, 4208. [Google Scholar] [CrossRef] [PubMed]
- Abal-Fabeiro, J.L.; Maside, X.; Bello, X.; Llovo, J.; Bartolome, C. Multilocus patterns of genetic variation across Cryptosporidium species suggest balancing selection at the gp60 locus. Mol. Ecol. 2013, 22, 4723–4732. [Google Scholar] [CrossRef]
- Feng, X.; Rich, S.M.; Akiyoshi, D.; Tumwine, J.K.; Kekitiinwa, A.; Nabukeera, N.; Tzipori, S.; Widmer, G. Extensive polymorphism in Cryptosporidium parvum identified by multilocus microsatellite analysis. Appl. Environ. Microbiol. 2000, 66, 3344–3349. [Google Scholar] [CrossRef]
- Tanriverdi, S.; Markovics, A.; Arslan, M.O.; Itik, A.; Shkap, V.; Widmer, G. Emergence of Distinct Genotypes of Cryptosporidium parvum in Structured Host Populations. Appl. Environ. Microbiol. 2006, 72, 2507–2513. [Google Scholar] [CrossRef]
- Tanriverdi, S.; Grinberg, A.; Chalmers, R.M.; Hunter, P.R.; Petrovic, Z.; Akiyoshi, D.E.; London, E.; Zhang, L.; Tzipori, S.; Tumwine, J.K.; et al. Inferences about the global population structure of Cryptosporidium parvum and Cryptosporidium hominis. Appl. Environ. Microbiol. 2008, 74, 7227–7234. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Tang, K.; Rowe, L.A.; Li, N.; Roellig, D.M.; Knipe, K.; Frace, M.; Yang, C.; Feng, Y.; Xiao, L. Comparative genomic analysis reveals occurrence of genetic recombination in virulent Cryptosporidium hominis subtypes and telomeric gene duplications in Cryptosporidium parvum. BMC Genom. 2015, 16, 320. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, N.; Roellig, D.M.; Kelley, A.; Liu, G.; Amer, S.; Tang, K.; Zhang, L.; Xiao, L. Comparative genomic analysis of the IId subtype family of Cryptosporidium parvum. Int. J. Parasitol. 2017, 47, 281–290. [Google Scholar] [CrossRef]
- Fei, J.; Wu, H.; Su, J.; Jin, C.; Li, N.; Guo, Y.; Feng, Y.; Xiao, L. Characterization of MEDLE-1, a protein in early development of Cryptosporidium parvum. Parasites Vectors 2018, 11, 312. [Google Scholar] [CrossRef]
- Li, B.; Wu, H.; Li, N.; Su, J.; Jia, R.; Jiang, J.; Feng, Y.; Xiao, L. Preliminary Characterization of MEDLE-2, a Protein Potentially Involved in the Invasion of Cryptosporidium parvum. Front. Microbiol. 2017, 8, 1647. [Google Scholar] [CrossRef]
- Su, J.; Jin, C.; Wu, H.; Fei, J.; Li, N.; Guo, Y.; Feng, Y.; Xiao, L. Differential Expression of Three Cryptosporidium Species-Specific MEDLE Proteins. Front. Microbiol. 2019, 10, 1177. [Google Scholar] [CrossRef] [PubMed]
- Gatei, W.; Barrett, D.; Lindo, J.F.; Eldemire-Shearer, D.; Cama, V.; Xiao, L. Unique Cryptosporidium population in HIV-infected persons, Jamaica. Emerg. Infect. Dis. 2008, 14, 841–843. [Google Scholar] [CrossRef]
- Widmer, G.; Tchack, L.; Spano, F.; Tzipori, S. A study of Cryptosporidium parvum genotypes and population structure. Mem. Inst. Oswaldo Cruz 1998, 93, 685–686. [Google Scholar] [CrossRef] [PubMed]
- Coupe, S.; Sarfati, C.; Hamane, S.; Derouin, F. Detection of Cryptosporidium and identification to the species level by nested PCR and restriction fragment length polymorphism. J. Clin. Microbiol. 2005, 43, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Leoni, F.; Mallon, M.E.; Smith, H.V.; Tait, A.; McLauchlin, J. Multilocus analysis of Cryptosporidium hominis and Cryptosporidium parvum isolates from sporadic and outbreak-related human cases and C. parvum isolates from sporadic livestock cases in the United Kingdom. J. Clin. Microbiol. 2007, 45, 3286–3294. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hunter, P.R.; Hadfield, S.J.; Wilkinson, D.; Lake, I.R.; Harrison, F.C.D.; Chalmers, R.M. Subtypes of Cryptosporidium parvum in humans and disease risk. Emerg. Infect. Dis. 2007, 13, 82–88. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef]
- Tanriverdi, S.; Arslan, M.O.; Akiyoshi, D.E.; Tzipori, S.; Widmer, G. Identification of genotypically mixed Cryptosporidium parvum populations in humans and calves. Mol. Biochem. Parasitol. 2003, 130, 13–22. [Google Scholar] [CrossRef]
- Kaupke, A.; Gawor, J.; Rzezutka, A.; Gromadka, R. Identification of pig-specific Cryptosporidium species in mixed infections using Illumina sequencing technology. Exp. Parasitol. 2017, 182, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Mercado, R.; Pena, S.; Ozaki, L.S.; Fredes, F.; Godoy, J. Multiple Cryptosporidium parvum subtypes detected in a unique isolate of a Chilean neonatal calf with diarrhea. Parasitol. Res. 2015, 114, 1985–1988. [Google Scholar] [CrossRef] [PubMed]
- Gan, M.; Liu, Q.; Yang, C.; Gao, Q.; Luo, T. Deep Whole-Genome Sequencing to Detect Mixed Infection of Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0159029. [Google Scholar] [CrossRef]
- Bones, A.J.; Josse, L.; More, C.; Miller, C.N.; Michaelis, M.; Tsaousis, A.D. Past and future trends of Cryptosporidium in vitro research. Exp. Parasitol. 2019, 196, 28–37. [Google Scholar] [CrossRef]
- Grinberg, A.; Widmer, G. Cryptosporidium within-host genetic diversity: Systematic bibliographical search and narrative overview. Int. J. Parasitol. 2016, 46, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Troell, K.; Hallstrom, B.; Divne, A.M.; Alsmark, C.; Arrighi, R.; Huss, M.; Beser, J.; Bertilsson, S. Cryptosporidium as a testbed for single cell genome characterization of unicellular eukaryotes. BMC Genom. 2016, 17, 471. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Rich, S.M.; Tzipori, S.; Widmer, G. Experimental evidence for genetic recombination in the opportunistic pathogen Cryptosporidium parvum. Mol. Biochem. Parasitol. 2002, 119, 55–62. [Google Scholar] [CrossRef]
- Yanta, C.A.; Bessonov, K.; Robinson, G.; Troell, K.; Guy, R.A. CryptoGenotyper: A new bioinformatics tool for rapid Cryptosporidium identification. Food Waterborne Parasitol. 2021, 23, e00115. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Zhu, S.J.; Almagro-Garcia, J.; McVean, G. Deconvolution of multiple infections in Plasmodium falciparum from high throughput sequencing data. Bioinformatics 2018, 34, 9–15. [Google Scholar] [CrossRef]
- Morada, M.; Lee, S.; Gunther-Cummins, L.; Weiss, L.M.; Widmer, G.; Tzipori, S.; Yarlett, N. Continuous culture of Cryptosporidium parvum using hollow fiber technology. Int. J. Parasitol. 2016, 46, 21–29. [Google Scholar] [CrossRef]
- Heo, I.; Dutta, D.; Schaefer, D.A.; Iakobachvili, N.; Artegiani, B.; Sachs, N.; Boonekamp, K.E.; Bowden, G.; Hendrickx, A.P.A.; Willems, R.J.L.; et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat. Microbiol. 2018, 3, 814–823. [Google Scholar] [CrossRef]
- DeCicco RePass, M.A.; Chen, Y.; Lin, Y.; Zhou, W.; Kaplan, D.L.; Ward, H.D. Novel Bioengineered Three-Dimensional Human Intestinal Model for Long-Term Infection of Cryptosporidium parvum. Infect. Immun. 2017, 85, e00731-16. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.; Funkhouser-Jones, L.J.; Wang, Y.; Ravindran, S.; Wang, Q.; Beatty, W.L.; Baldridge, M.T.; VanDussen, K.L.; Shen, B.; Kuhlenschmidt, M.S.; et al. A Stem-Cell-Derived Platform Enables Complete Cryptosporidium Development In Vitro and Genetic Tractability. Cell Host Microbe 2019, 26, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Yarlett, N.; Morada, M.; Gobin, M.; Van Voorhis, W.; Arnold, S. In Vitro Culture of Cryptosporidium parvum Using Hollow Fiber Bioreactor: Applications for Simultaneous Pharmacokinetic and Pharmacodynamic Evaluation of Test Compounds. Methods Mol. Biol. 2020, 2052, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Navin, N.E. Advances and applications of single-cell sequencing technologies. Mol. Cell 2015, 58, 598–609. [Google Scholar] [CrossRef]
- Anusz, K.Z.; Mason, P.H.; Riggs, M.W.; Perryman, L.E. Detection of Cryptosporidium parvum oocysts in bovine feces by monoclonal antibody capture enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1990, 28, 2770–2774. [Google Scholar] [CrossRef]
- Lee, S.; Beamer, G.; Tzipori, S. The piglet acute diarrhea model for evaluating efficacy of treatment and control of cryptosporidiosis. Hum. Vaccines Immunother. 2019, 15, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Riggs, M.W.; Schaefer, D.A. Calf Clinical Model of Cryptosporidiosis for Efficacy Evaluation of Therapeutics. Methods Mol. Biol. 2020, 2052, 253–282. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.R.; Healey, M.C. Experimental Cryptosporidium parvum infections in immunosuppressed adult mice. Infect. Immun. 1992, 60, 1648–1652. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.; Galinsky, K.; Rogov, P.; Fennell, T.; Van Tyne, D.; Russ, C.; Daniels, R.; Barnes, K.G.; Bochicchio, J.; Ndiaye, D.; et al. Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 2011, 12, R73. [Google Scholar] [CrossRef] [PubMed]
- Hupalo, D.N.; Luo, Z.; Melnikov, A.; Sutton, P.L.; Rogov, P.; Escalante, A.; Vallejo, A.F.; Herrera, S.; Arevalo-Herrera, M.; Fan, Q.; et al. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat. Genet. 2016, 48, 953–958. [Google Scholar] [CrossRef]
- Khan, A.; Ferreira, E.C.A.; Grigg, M.E. Development of SureSelect Target Enrichment for Whole Genome Sequencing of Cryptosporidium Directly from Stool Samples. Available online: https://en.rouentourisme.com/wp-content/uploads//2019/06/Programme-définitif-modifié-200619.pdf (p. 99) and https://hal-normandie-univ.archives-ouvertes.fr/hal-02495405 (accessed on 6 March 2021).
- Kissinger, J.C.; Glenn, T.C. Capturing the Genomic Variation Present in Cryptosporidium and Cryptosporidiosis (1R01AI148667-01A1). Available online: https://reporter.nih.gov/project-details/10053025 (accessed on 6 March 2021).
- CryptoCapture.org: A Large Community Effort to Survey the Population Genetic Structure of Human-Infecting Cryptosporidia. Available online: http://cryptocapture.org/ (accessed on 6 March 2021).


{kind=link}
| Cryptosporidium Species | # of Genome Sequences Available | Sequencing Technology | Gene Evidence Availability | |||
|---|---|---|---|---|---|---|
| RNAseq a | Expressed Sequence Tag Datasets | Proteomic Data | # of Genome Annotations Available | |||
| C. parvum | 19 | Sanger, Illumina, 454, ABI SOLiD, PacBio, ONT, HAPPY map data | Yes | Yes | Yes | 2 |
| C. hominis | 12 | Sanger, Illumina, Ion Torrent, 454 | Yes | Yes | Yes | 5 |
| C. ubiquitum | 5 | Illumina | No | No | No | 1 |
| C. meleagridis | 3 | Illumina | No | No | No | 1 |
| C. andersoni | 3 | Illumina | No | No | No | 1 |
| C. muris | 1 | Sanger and 454 | No | No | Yes | 1 |
| C. tyzzeri | 1 | Illumina | No | No | No | 1 |
| C. felis | 1 | Illumina | No | No | No | 1 |
| C. cuniculus | 1 | Illumina | No | No | No | 0 |
| C. ryanae | 1 | Illumina | No | No | No | 0 |
| C. bovis | 1 | Illumina | No | No | No | 0 |
| C. viatorum | 1 | Illumina | No | No | No | 0 |
| C. sp. 37763 | 1 | Illumina | No | No | No | 0 |
| C. sp. chipmunk LX-2015 | 1 | Illumina | No | No | No | 0 |
| C. baileyi | 1 | Illumina, PacBio | Yes | Yes | No | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baptista, R.P.; Cooper, G.W.; Kissinger, J.C. Challenges for Cryptosporidium Population Studies. Genes 2021, 12, 894. https://doi.org/10.3390/genes12060894
Baptista RP, Cooper GW, Kissinger JC. Challenges for Cryptosporidium Population Studies. Genes. 2021; 12(6):894. https://doi.org/10.3390/genes12060894
Chicago/Turabian StyleBaptista, Rodrigo P., Garrett W. Cooper, and Jessica C. Kissinger. 2021. "Challenges for Cryptosporidium Population Studies" Genes 12, no. 6: 894. https://doi.org/10.3390/genes12060894
APA StyleBaptista, R. P., Cooper, G. W., & Kissinger, J. C. (2021). Challenges for Cryptosporidium Population Studies. Genes, 12(6), 894. https://doi.org/10.3390/genes12060894