
The Diagnostic Journey of a Patient with Prader–Willi-Like Syndrome and a Unique Homozygous SNURF-SNRPN Variant; Bio-Molecular Analysis and Review of the Literature

 ,
,  ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Methylation-Specific Multiplex-Ligation Probe Amplification (MS-MPLA)
2.2. Single Nucleotide Polymorphism (SNP) Array
2.3. Next Generation Sequencing (NSG)
2.4. RNA Sequencing
2.5. Effect Predictor Programs
2.6. PRiSM Screen
2.6.1. Constructs
2.6.2. Mice
2.6.3. HEK-293T Cell Transfections
2.6.4. Western Blot
2.6.5. Primary Hippocampal Cultures
2.6.6. Neuronal Transfection and Immunocytochemistry
2.6.7. In Utero Electroporation
2.6.8. Immunohistochemistry
2.6.9. Statistical Analysis
2.7. Literature Review
3. Results
3.1. Patient Description
3.1.1. Medical History
3.1.2. Anamnesis
3.1.3. Family History
3.1.4. Physical Examination
3.1.5. Biochemistry and Imaging
3.1.6. Polysomnography
3.1.7. Features Not Corresponding to PWS
3.2. Literature Review
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Holland, A.J.; Hauffa, B.P.; Hokken-Koelega, A.C.; Tauber, M. Recommendations for the diagnosis and management of prader-willi syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4183–4197. [Google Scholar] [CrossRef]
- Pellikaan, K.; Rosenberg, A.G.W.; Kattentidt-Mouravieva, A.A.; Kersseboom, R.; Bos-Roubos, A.G.; Veen-Roelofs, J.M.C.; van Wieringen, N.; Hoekstra, F.M.E.; van den Berg, S.A.A.; van der Lely, A.J.; et al. Missed diagnoses and health problems in adults with prader-willi syndrome: Recommendations for screening and treatment. J. Clin. Endocrinol. Metab. 2020, 105, e4671–e4687. [Google Scholar] [CrossRef]
- Williams, M.S.; Rooney, B.L.; Williams, J.; Josephson, K.; Pauli, R. Investigation of thermoregulatory characteristics in patients with prader-willi syndrome. Am. J. Med. Genet. 1994, 49, 302–307. [Google Scholar] [CrossRef]
- Wattendorf, D.J.; Muenke, M. Prader-willi syndrome. Am. Fam. Physician 2005, 72, 827–830. [Google Scholar] [PubMed]
- Bennett, J.A.; Germani, T.; Haqq, A.M.; Zwaigenbaum, L. Autism spectrum disorder in prader-willi syndrome: A systematic review. Am. J. Med. Genet. A 2015, 167A, 2936–2944. [Google Scholar] [CrossRef] [PubMed]
- Holm, V.A.; Cassidy, S.B.; Butler, M.G.; Hanchett, J.M.; Greenswag, L.R.; Whitman, B.Y.; Greenberg, F. Prader-willi syndrome: Consensus diagnostic criteria. Pediatrics 1993, 91, 398–402. [Google Scholar]
- Buiting, K.; Cassidy, S.B.; Driscoll, D.J.; Gillessen-Kaesbach, G.; Kanber, D.; Tauber, M.; Schwinger, E.; Horsthemke, B. Clinical utility gene card for: Prader-willi syndrome. Eur. J. Hum. Genet. 2014, 22, 1153. [Google Scholar] [CrossRef]
- Cheon, C.K. Genetics of prader-willi syndrome and Prader-Will-like syndrome. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.F.; Paiva, C.L. Prader-Willi-like phenotypes: A systematic review of their chromosomal abnormalities. Genet. Mol. Res. 2014, 13, 2290–2298. [Google Scholar] [CrossRef] [PubMed]
- Desch, L.; Marle, N.; Mosca-Boidron, A.L.; Faivre, L.; Eliade, M.; Payet, M.; Ragon, C.; Thevenon, J.; Aral, B.; Ragot, S.; et al. 6q16.3q23.3 duplication associated with Prader-Willi-like syndrome. Mol. Cytogenet. 2015, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Potter, K.J.; Burnett, L.C.; Orsso, C.E.; Inman, M.; Ryman, D.C.; Haqq, A.M. Prader–Willi-like phenotype caused by an atypical 15q11.2 microdeletion. Genes 2020, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; Del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the hbii-85 c/d box small nucleolar rna cluster. Nat. Genet. 2008, 40, 719–721. [Google Scholar] [CrossRef]
- De Smith, A.J.; Purmann, C.; Walters, R.G.; Ellis, R.J.; Holder, S.E.; Van Haelst, M.M.; Brady, A.F.; Fairbrother, U.L.; Dattani, M.; Keogh, J.M.; et al. A deletion of the hbii-85 class of small nucleolar rnas (snornas) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 2009, 18, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Duker, A.L.; Ballif, B.C.; Bawle, E.V.; Person, R.E.; Mahadevan, S.; Alliman, S.; Thompson, R.; Traylor, R.; Bejjani, B.A.; Shaffer, L.G.; et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the snord116 c/d box snorna cluster in prader-willi syndrome. Eur. J. Hum. Genet. 2010, 18, 1196–1201. [Google Scholar] [CrossRef]
- Bieth, E.; Eddiry, S.; Gaston, V.; Lorenzini, F.; Buffet, A.; Conte Auriol, F.; Molinas, C.; Cailley, D.; Rooryck, C.; Arveiler, B.; et al. Highly restricted deletion of the snord116 region is implicated in prader-willi syndrome. Eur. J. Hum. Genet. 2015, 23, 252–255. [Google Scholar] [CrossRef]
- Hassan, M.; Butler, M.G. Prader-Willi syndrome and atypical submicroscopic 15q11-q13 deletions with or without imprinting defects. Eur. J. Med. Genet. 2016, 59, 584–589. [Google Scholar] [CrossRef]
- Fontana, P.; Grasso, M.; Acquaviva, F.; Gennaro, E.; Galli, M.L.; Falco, M.; Scarano, F.; Scarano, G.; Lonardo, F. Snord116 deletions cause prader-willi syndrome with a mild phenotype and macrocephaly. Clin. Genet. 2017, 92, 440–443. [Google Scholar] [CrossRef]
- Beygo, J.; Buiting, K.; Ramsden, S.C.; Ellis, R.; Clayton-Smith, J.; Kanber, D. Update of the emqn/acgs best practice guidelines for molecular analysis of prader-willi and angelman syndromes. Eur. J. Hum. Genet. 2019, 27, 1326–1340. [Google Scholar] [CrossRef]
- Srebniak, M.; Boter, M.; Oudesluijs, G.; Joosten, M.; Govaerts, L.; Van Opstal, D.; Galjaard, R.J. Application of snp array for rapid prenatal diagnosis: Implementation, genetic counselling and diagnostic flow. Eur. J. Hum. Genet. 2011, 19, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Kleinendorst, L.; Massink, M.P.G.; Cooiman, M.I.; Savas, M.; van der Baan-Slootweg, O.H.; Roelants, R.J.; Janssen, I.C.M.; Meijers-Heijboer, H.J.; Knoers, N.; Ploos van Amstel, H.K.; et al. Genetic obesity: Next-generation sequencing results of 1230 patients with obesity. J. Med. Genet. 2018, 55, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive statistical study of 452 brca1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. Sift missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. Sift web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using polyphen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Van Woerden, G.M.; Bos, M.; de Konink, C.; Distel, B.; Avagliano Trezza, R.; Shur, N.E.; Barañano, K.; Mahida, S.; Chassevent, A.; Schreiber, A.; et al. Taok1 is associated with neurodevelopmental disorder and essential for neuronal maturation and cortical development. Hum. Mutat. 2021, 42, 445–459. [Google Scholar] [CrossRef]
- Heimer, G.; van Woerden, G.M.; Barel, O.; Marek-Yagel, D.; Kol, N.; Munting, J.B.; Borghei, M.; Atawneh, O.M.; Nissenkorn, A.; Rechavi, G.; et al. Netrin-g2 dysfunction causes a rett-like phenotype with areflexia. Hum. Mutat. 2020, 41, 476–486. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Kousi, M.; van Woerden, G.M.; Klein, M.; Bralten, J.; Mancini, G.M.S.; van Essen, T.; Proietti-Onori, M.; Smeets, E.E.J.; van Gastel, M.; et al. Variation in a range of mtor-related genes associates with intracranial volume and intellectual disability. Nat. Commun. 2017, 8, 1052. [Google Scholar] [CrossRef]
- Küry, S.; van Woerden, G.M.; Besnard, T.; Proietti Onori, M.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De novo mutations in protein kinase genes camk2a and camk2b cause intellectual disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef]
- Proietti Onori, M.; Koopal, B.; Everman, D.B.; Worthington, J.D.; Jones, J.R.; Ploeg, M.A.; Mientjes, E.; van Bon, B.W.; Kleefstra, T.; Schulman, H.; et al. The intellectual disability-associated camk2g p.Arg292pro mutation acts as a pathogenic gain-of-function. Hum. Mutat. 2018, 39, 2008–2024. [Google Scholar] [CrossRef]
- Goslin, K.; Banker, G. Culturing Nerve Cells; MIT Press: Cambridge, MA, USA, 1991. [Google Scholar]
- Fredriks, A.M.; van Buuren, S.; Burgmeijer, R.J.F.; Meulmeester, J.F.; Beuker, R.J.; Brugman, E.; Roede, M.J.; Verloove-Vanhorick, S.P.; Wit, J.-M. Continuing positive secular growth change in the netherlands 1955–1997. Pediatr. Res. 2000, 47, 316–323. [Google Scholar] [CrossRef]
- Corneli, G.; Di Somma, C.; Baldelli, R.; Rovere, S.; Gasco, V.; Croce, C.G.; Grottoli, S.; Maccario, M.; Colao, A.; Lombardi, G.; et al. The cut-off limits of the gh response to gh-releasing hormone-arginine test related to body mass index. Eur. J. Endocrinol. 2005, 153, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Ruehland, W.R.; Rochford, P.D.; O’Donoghue, F.J.; Pierce, R.J.; Singh, P.; Thornton, A.T. The new aasm criteria for scoring hypopneas: Impact on the apnea hypopnea index. Sleep 2009, 32, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Kelberman, D.; Dattani, M.T. Genetics of septo-optic dysplasia. Pituitary 2007, 10, 393–407. [Google Scholar] [CrossRef]
- Fintini, D.; Pedicelli, S.; Bocchini, S.; Bizzarri, C.; Grugni, G.; Cappa, M.; Crinò, A. 25oh vitamin d levels in pediatric patients affected by prader–willi syndrome. J. Endocrinol. Investig. 2018, 41, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, L.; Joensson, I.M.; Froekjaer, J.B.; Krogh, K.; Farholt, S. A descriptive study of colorectal function in adults with prader-willi syndrome: High prevalence of constipation. BMC Gastroenterol. 2014, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Sinnema, M.; Maaskant, M.A.; van Schrojenstein Lantman-de Valk, H.M.; van Nieuwpoort, I.C.; Drent, M.L.; Curfs, L.M.; Schrander-Stumpel, C.T. Physical health problems in adults with prader-willi syndrome. Am. J. Med. Genet. A 2011, 155A, 2112–2124. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.J.; Lapcevic, R.; Romero, A.E.; Yoon, M.; Das, J.; Beltrán, J.F.; Mort, M.; Stenson, P.D.; Cooper, D.N.; Paccanaro, A.; et al. Mutation3d: Cancer gene prediction through atomic clustering of coding variants in the structural proteome. Hum. Mutat. 2016, 37, 447–456. [Google Scholar] [CrossRef]
- Rossi, E.; Giorda, R.; Bonaglia, M.C.; Di Candia, S.; Grechi, E.; Franzese, A.; Soli, F.; Rivieri, F.; Patricelli, M.G.; Saccilotto, D.; et al. De novo unbalanced translocations in Prader-Willi and angelman syndrome might be the reciprocal product of inv dup(15)s. PLoS ONE 2012, 7, e39180. [Google Scholar] [CrossRef] [PubMed]
- Gunay-Aygun, M.; Schwartz, S.; Heeger, S.; O’Riordan, M.A.; Cassidy, S.B. The changing purpose of prader-willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 2001, 108, E92. [Google Scholar] [CrossRef] [PubMed]
- Greger, V.; Woolf, E.; Lalande, M. Cloning of the breakpoints of a submicroscopic deletion in an angelman syndrome patient. Hum. Mol. Genet. 1993, 2, 921–924. [Google Scholar] [CrossRef]
- Hamabe, J.I.; Kuroki, Y.; Imaizumi, K.; Sugimoto, T.; Fukushima, Y.; Yamaguchi, A.; Izumikawa, Y.; Niikawa, N. DNA deletion and its parental origin in angelman syndrome patients. Am. J. Med. Genet. 1991, 41, 64–68. [Google Scholar] [CrossRef]
- Schulze, A.; Hansen, C.; Skakkebaek, N.E.; Brondum-Nielsen, K.; Ledbeter, D.H.; Tommerup, N. Exclusion of snrpn as a major determinant of prader-willi syndrome by a translocation breakpoint. Nat. Genet. 1996, 12, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Nicholls, R.D.; Butler, M.G.; Saitoh, S.; Hainline, B.E.; Palmer, C.G. Breakage in the snrpn locus in a balanced 46,xy,t(15;19) prader-willi syndrome patient. Hum. Mol. Genet. 1996, 5, 517–524. [Google Scholar] [CrossRef]
- Conroy, J.M.; Grebe, T.A.; Becker, L.A.; Tsuchiya, K.; Nicholls, R.D.; Buiting, K.; Horsthemke, B.; Cassidy, S.B.; Schwartz, S. Balanced translocation 46,xy,t(2;15)(q37.2;q11.2) associated with atypical prader-willi syndrome. Am. J. Hum. Genet. 1997, 61, 388–394. [Google Scholar] [CrossRef][Green Version]
- Kuslich, C.D.; Kobori, J.A.; Mohapatra, G.; Gregorio-King, C.; Donlon, T.A. Prader-Willi syndrome is caused by disruption of the snrpn gene. Am. J. Hum. Genet. 1999, 64, 70–76. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wirth, J.; Back, E.; Hüttenhofer, A.; Nothwang, H.G.; Lich, C.; Groß, S.; Menzel, C.; Schinzel, A.; Kioschis, P.; Tommerup, N.; et al. A translocation breakpoint cluster disrupts the newly defined 3′ end of the snurf-snrpn transcription unit on chromosome 15. Hum. Mol. Genet. 2001, 10, 201–210. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maina, E.N.; Webb, T.; Soni, S.; Whittington, J.; Boer, H.; Clarke, D.; Holland, A. Analysis of candidate imprinted genes in pws subjects with atypical genetics: A possible inactivating mutation in the snurf/snrpn minimal promoter. J. Hum. Genet. 2007, 52, 297–307. [Google Scholar] [CrossRef]
- Whittington, J.; Holland, A.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Relationship between clinical and genetic diagnosis of prader-willi syndrome. J. Med. Genet. 2002, 39, 926–932. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bürger, J.; Horn, D.; Tönnies, H.; Neitzel, H.; Reis, A. Familial interstitial 570 kbp deletion of the ube3a gene region causing angelman syndrome but not prader-willi syndrome. Am. J. Med. Genet. 2002, 111, 233–237. [Google Scholar] [CrossRef]
- Runte, M.; Varon, R.; Horn, D.; Horsthemke, B.; Buiting, K. Exclusion of the c/d box snorna gene cluster hbii-52 from a major role in prader-willi syndrome. Hum. Genet. 2005, 116, 228–230. [Google Scholar] [CrossRef]
- Schüle, B.; Albalwi, M.; Northrop, E.; Francis, D.I.; Rowell, M.; Slater, H.R.; Gardner, R.J.; Francke, U. Molecular breakpoint cloning and gene expression studies of a novel translocation t(4;15)(q27;q11.2) associated with prader-willi syndrome. BMC Med. Genet. 2005, 6, 18. [Google Scholar] [CrossRef]
- Calounova, G.; Hedvicakova, P.; Silhanova, E.; Kreckova, G.; Sedlacek, Z. Molecular and clinical characterization of two patients with prader-willi syndrome and atypical deletions of proximal chromosome 15q. Am. J. Med. Genet. Part A 2008, 146, 1955–1962. [Google Scholar] [CrossRef]
- Kanber, D.; Giltay, J.; Wieczorek, D.; Zogel, C.; Hochstenbach, R.; Caliebe, A.; Kuechler, A.; Horsthemke, B.; Buiting, K. A paternal deletion of mkrn3, magel2 and ndn does not result in prader-willi syndrome. Eur. J. Hum. Genet. 2009, 17, 582–590. [Google Scholar] [CrossRef]
- Naik, S.; Thomas, N.S.; Davies, J.H.; Lever, M.; Raponi, M.; Baralle, D.; Temple, I.K.; Caliebe, A. Novel tandem duplication in exon 1 of the snurf/snrpn gene in a child with transient excessive eating behaviour and weight gain. Mol. Syndr. 2011, 2, 76–80. [Google Scholar] [CrossRef]
- Rivera, H.; Zuffardi, O.; Gargantini, L. Nonreciprocal and jumping translocations of 15q1----qter in prader-willi syndrome. Am J. Med. Genet. 1990, 37, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Fraccaro, M.; Zuffardi, O.; Buhler, E. Deficiency, transposition, and duplication of one 15q region may be alternatively associated with prader-willi (or a similar) syndrome. Analysis of seven cases after varying ascertainment. Hum. Genet. 1983, 64, 388–394. [Google Scholar] [CrossRef]
- Buiting, K.; Di Donato, N.; Beygo, J.; Bens, S.; von der Hagen, M.; Hackmann, K.; Horsthemke, B. Clinical phenotypes of magel2 mutations and deletions. Orphanet J. Rare Dis. 2014, 9, 40. [Google Scholar] [CrossRef]
- Koufaris, C.; Alexandrou, A.; Papaevripidou, I.; Alexandrou, I.; Christophidou-Anastasiadou, V.; Sismani, C. Deletion of snurf/snrpn u1b and u1b* upstream exons in a child with developmental delay and excessive weight. J. Genet. 2016, 95, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Mitsuhashi, S.; Miyake, N.; Ohta, T.; Liang, D.; Wu, L.; Matsumoto, N. Translocation breakpoint disrupting the host snhg14 gene but not coding genes or snornas in typical prader-willi syndrome. J. Hum. Genet. 2019, 64, 647–652. [Google Scholar] [CrossRef]
- Yamada, K.; Matsuzawa, H.; Uchiyama, M.; Kwee, I.L.; Nakada, T. Brain developmental abnormalities in prader-willi syndrome detected by diffusion tensor imaging. Pediatrics 2006, 118, e442–e448. [Google Scholar] [CrossRef] [PubMed]
- Azor, A.M.; Cole, J.H.; Holland, A.J.; Dumba, M.; Patel, M.C.; Sadlon, A.; Goldstone, A.P.; Manning, K.E. Increased brain age in adults with prader-willi syndrome. Neuroimage Clin. 2019, 21, 101664. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, Y.; von Deneen, K.M.; Zhu, H.; Gao, J.H. Brain structural alterations in obese children with and without prader-willi syndrome. Hum. Brain Mapp. 2017, 38, 4228–4238. [Google Scholar] [CrossRef]
- Lukoshe, A.; White, T.; Schmidt, M.N.; van der Lugt, A.; Hokken-Koelega, A.C. Divergent structural brain abnormalities between different genetic subtypes of children with prader-willi syndrome. J. Neurodev. Disord. 2013, 5, 31. [Google Scholar] [CrossRef]
- Miller, J.L.; Couch, J.A.; Schmalfuss, I.; He, G.; Liu, Y.; Driscoll, D.J. Intracranial abnormalities detected by three-dimensional magnetic resonance imaging in prader–willi syndrome. Am. J. Med. Genet. Part A 2007, 143A, 476–483. [Google Scholar] [CrossRef]
- Miller, S.P.; Shevell, M.I.; Patenaude, Y.; Poulin, C.; O’Gorman, A.M. Septo-optic dysplasia plus: A spectrum of malformations of cortical development. Neurology 2000, 54, 1701–1703. [Google Scholar] [CrossRef]
- Polizzi, A.; Pavone, P.; Iannetti, P.; Manfré, L.; Ruggieri, M. Septo-optic dysplasia complex: A heterogeneous malformation syndrome. Pediatr. Neurol. 2006, 34, 66–71. [Google Scholar] [CrossRef] [PubMed]
- McNay, D.E.; Turton, J.P.; Kelberman, D.; Woods, K.S.; Brauner, R.; Papadimitriou, A.; Keller, E.; Keller, A.; Haufs, N.; Krude, H.; et al. Hesx1 mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J. Clin. Endocrinol. Metab. 2007, 92, 691–697. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.J.; Alatzoglou, K.S.; Dattani, M.T. Septo-optic dysplasia and other midline defects: The role of transcription factors: Hesx1 and beyond. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Houtkoop, W. Basisvaardigheden in Nederland; Max Goote Kenniscentrum: Amsterdam, The Netherlands, 1999; pp. 1–196. [Google Scholar]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Gray, T.A.; Saitoh, S.; Nicholls, R.D. An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 5616–5621. [Google Scholar] [CrossRef]
- Glenn, C.C.; Saitoh, S.; Jong, M.T.; Filbrandt, M.M.; Surti, U.; Driscoll, D.J.; Nicholls, R.D. Gene structure, DNA methylation, and imprinted expression of the human snrpn gene. Am. J. Hum. Genet. 1996, 58, 335–346. [Google Scholar] [PubMed]
- Schmauss, C.; Brines, M.L.; Lerner, M.R. The gene encoding the small nuclear ribonucleoprotein-associated protein n is expressed at high levels in neurons. J. Biol. Chem. 1992, 267, 8521–8529. [Google Scholar] [CrossRef]
- Ozcelik, T.; Leff, S.; Robinson, W.; Donlon, T.; Lalande, M.; Sanjines, E.; Schinzel, A.; Francke, U. Small nuclear ribonucleoprotein polypeptide n (snrpn), an expressed gene in the prader-willi syndrome critical region. Nat. Genet. 1992, 2, 265–269. [Google Scholar] [CrossRef]
- Li, H.; Zhao, P.; Xu, Q.; Shan, S.; Hu, C.; Qiu, Z.; Xu, X. The autism-related gene snrpn regulates cortical and spine development via controlling nuclear receptor nr4a1. Sci. Rep. 2016, 6, 29878. [Google Scholar] [CrossRef]
- Runte, M.; Huttenhofer, A.; Gross, S.; Kiefmann, M.; Horsthemke, B.; Buiting, K. The ic-snurf-snrpn transcript serves as a host for multiple small nucleolar rna species and as an antisense rna for ube3a. Hum. Mol. Genet. 2001, 10, 2687–2700. [Google Scholar] [CrossRef]
- Gallagher, R.C.; Pils, B.; Albalwi, M.; Francke, U. Evidence for the role of pwcr1/hbii-85 c/d box small nucleolar rnas in prader-willi syndrome. Am. J. Hum. Genet. 2002, 71, 669–678. [Google Scholar] [CrossRef]
- Dittrich, B.; Buiting, K.; Korn, B.; Rickard, S.; Buxton, J.; Saitoh, S.; Nicholls, R.D.; Poustka, A.; Winterpacht, A.; Zabel, B.; et al. Imprint switching on human chromosome 15 may involve alternative transcripts of the snrpn gene. Nat. Genet. 1996, 14, 163–170. [Google Scholar] [CrossRef]
- Farber, C.; Dittrich, B.; Buiting, K.; Horsthemke, B. The chromosome 15 imprinting centre (ic) region has undergone multiple duplication events and contains an upstream exon of snrpn that is deleted in all angelman syndrome patients with an ic microdeletion. Hum. Mol. Genet. 1999, 8, 337–343. [Google Scholar] [CrossRef][Green Version]
- Buiting, K.; Saitoh, S.; Gross, S.; Dittrich, B.; Schwartz, S.; Nicholls, R.D.; Horsthemke, B. Inherited microdeletions in the angelman and prader-willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 1995, 9, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Blaydes, S.M.; Elmore, M.; Yang, T.; Brannan, C.I. Analysis of murine snrpn and human snrpn gene imprinting in transgenic mice. Mamm. Genome 1999, 10, 549–555. [Google Scholar] [CrossRef]
- Horsthemke, B.; Buiting, K. Imprinting defects on human chromosome 15. Cytogenet. Genome Res. 2006, 113, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Gray, T.A.; Rogan, P.K.; Buiting, K.; Gabriel, J.M.; Saitoh, S.; Muralidhar, B.; Bilienska, B.; Krajewska-Walasek, M.; Driscoll, D.J.; et al. Imprinting-mutation mechanisms in prader-willi syndrome. Am. J. Hum. Genet. 1999, 64, 397–413. [Google Scholar] [CrossRef]
- Rabinovitz, S.; Kaufman, Y.; Ludwig, G.; Razin, A.; Shemer, R. Mechanisms of activation of the paternally expressed genes by the prader-willi imprinting center in the prader-willi/angelman syndromes domains. Proc. Natl. Acad. Sci. USA 2012, 109, 7403–7408. [Google Scholar] [CrossRef] [PubMed]
- Perk, J.; Makedonski, K.; Lande, L.; Cedar, H.; Razin, A.; Shemer, R. The imprinting mechanism of the prader-willi/angelman regional control center. EMBO J. 2002, 21, 5807–5814. [Google Scholar] [CrossRef]
- Horsthemke, B.; Wagstaff, J. Mechanisms of imprinting of the prader-willi/angelman region. Am. J. Med. Genet. A 2008, 146A, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Nakao, M.; Christian, S.; Örstavik, K.H.; Tommerup, N.; Ledbetter, D.H.; Beaudet, A.L. Deletions of a differentially methylated cpg island at the snrpn gene define a putative imprinting control region. Nat. Genet. 1994, 8, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.D.; Saitoh, S.; Horsthemke, B. Imprinting in prader-willi and angelman syndromes. Trends Genet. 1998, 14, 194–200. [Google Scholar] [CrossRef]
- McCarthy, J.; Lupo, P.J.; Kovar, E.; Rech, M.; Bostwick, B.; Scott, D.; Kraft, K.; Roscioli, T.; Charrow, J.; Schrier Vergano, S.A.; et al. Schaaf-yang syndrome overview: Report of 78 individuals. Am. J. Med. Genet. A 2018, 176, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- Kleinendorst, L.; Pi Castán, G.; Caro-Llopis, A.; Boon, E.M.J.; van Haelst, M.M. The role of obesity in the fatal outcome of schaaf-yang syndrome: Early onset morbid obesity in a patient with a magel2 mutation. Am. J. Med. Genet. A 2018, 176, 2456–2459. [Google Scholar] [CrossRef]
- Shen, M.; Eyras, E.; Wu, J.; Khanna, A.; Josiah, S.; Rederstorff, M.; Zhang, M.Q.; Stamm, S. Direct cloning of double-stranded rnas from rnase protection analysis reveals processing patterns of c/d box snornas and provides evidence for widespread antisense transcript expression. Nucleic Acids Res. 2011, 39, 9720–9730. [Google Scholar] [CrossRef] [PubMed]
- Wevrick, R.; Francke, U. An imprinted mouse transcript homologous to the human imprinted in prader-willi syndrome (ipw) gene. Hum. Mol. Genet. 1997, 6, 325–332. [Google Scholar] [CrossRef]
- Wevrick, R.; Kerns, J.A.; Francke, U. Identification of a novel paternally expressed gene in the prader-willi syndrome region. Hum. Mol. Genet. 1994, 3, 1877–1882. [Google Scholar] [CrossRef]
- Adhikari, A.; Copping, N.A.; Onaga, B.; Pride, M.C.; Coulson, R.L.; Yang, M.; Yasui, D.H.; LaSalle, J.M.; Silverman, J.L. Cognitive deficits in the snord116 deletion mouse model for prader-willi syndrome. Neurobiol. Learn. Mem. 2019, 165, 106874. [Google Scholar] [CrossRef]
- Ding, F.; Li, H.H.; Zhang, S.; Solomon, N.M.; Camper, S.A.; Cohen, P.; Francke, U. Snorna snord116 (pwcr1/mbii-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 2008, 3, e1709. [Google Scholar] [CrossRef]
- Tsai, T.F.; Jiang, Y.H.; Bressler, J.; Armstrong, D.; Beaudet, A.L. Paternal deletion from snrpn to ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to prader-willi syndrome. Hum. Mol. Genet. 1999, 8, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Adamson, T.E.; Resnick, J.L.; Leff, S.; Wevrick, R.; Francke, U.; Jenkins, N.A.; Copeland, N.G.; Brannan, C.I. A mouse model for prader-willi syndrome imprinting-centre mutations. Nat. Genet. 1998, 19, 25–31. [Google Scholar] [CrossRef]
- Rieusset, A.; Schaller, F.; Unmehopa, U.; Matarazzo, V.; Watrin, F.; Linke, M.; Georges, B.; Bischof, J.; Dijkstra, F.; Bloemsma, M.; et al. Stochastic loss of silencing of the imprinted ndn/ndn allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. PLoS Genet. 2013, 9, e1003752. [Google Scholar] [CrossRef]
- Chamberlain, S.J.; Johnstone, K.A.; DuBose, A.J.; Simon, T.A.; Bartolomei, M.S.; Resnick, J.L.; Brannan, C.I. Evidence for genetic modifiers of postnatal lethality in pws-ic deletion mice. Hum. Mol. Genet. 2004, 13, 2971–2977. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Korostelev, S.A.; Zelenova, M.A.; Yurov, Y.B. Long contiguous stretches of homozygosity spanning shortly the imprinted loci are associated with intellectual disability, autism and/or epilepsy. Mol. Cytogenet. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gray, T.A.; Smithwick, M.J.; Schaldach, M.A.; Martone, D.L.; Graves, J.A.; McCarrey, J.R.; Nicholls, R.D. Concerted regulation and molecular evolution of the duplicated snrpb’/b and snrpn loci. Nucleic Acids Res. 1999, 27, 4577–4584. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
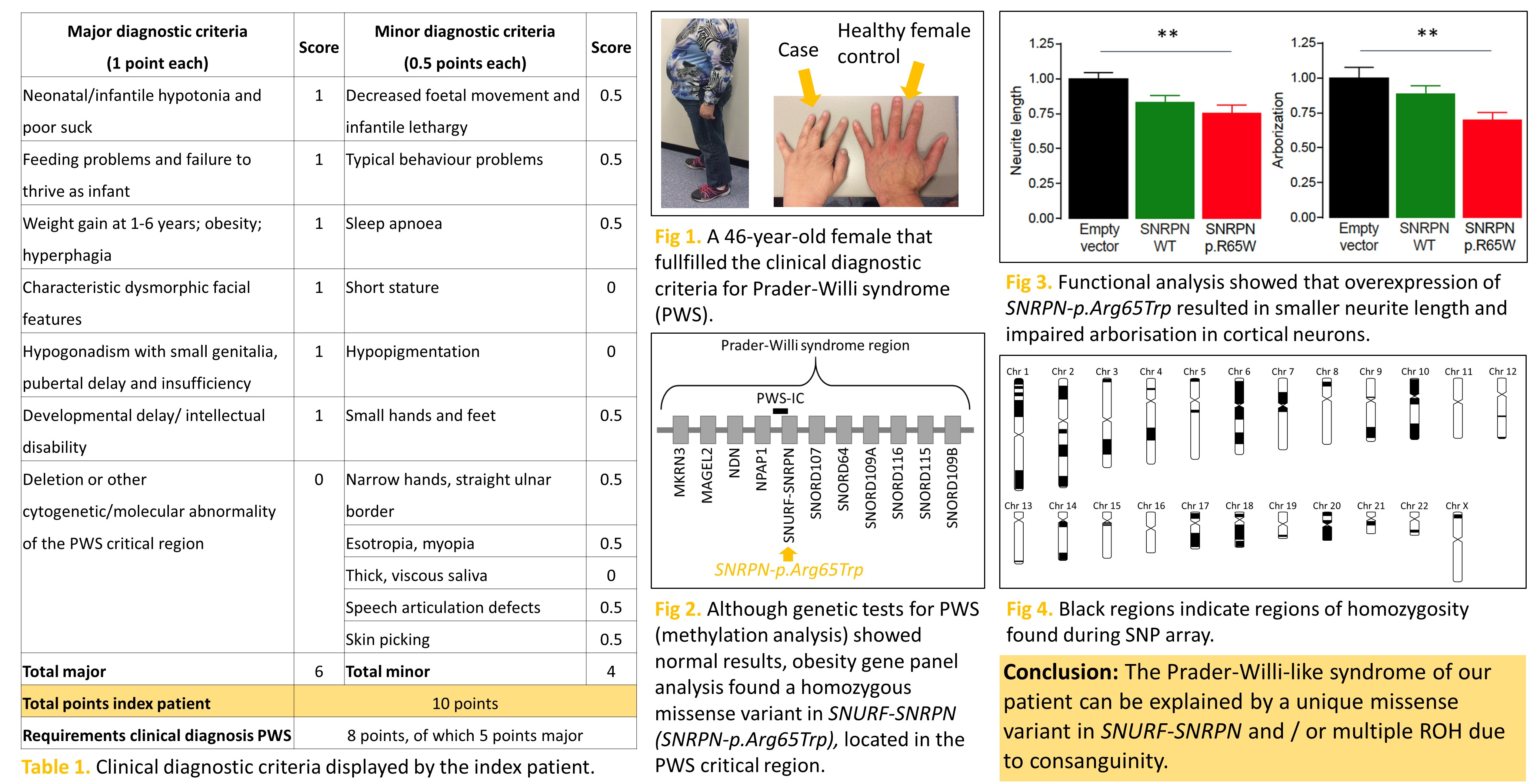
| Major Criteria (1 Point Each) | Score | Minor Criteria (0.5 Points Each) | Score |
|---|---|---|---|
| Neonatal/infantile hypotonia and poor suck | 1 | Decreased foetal movement and infantile lethargy | 0.5 |
| Feeding problems and failure to thrive as infant | 1 | Typical behaviour problems | 0.5 |
| Weight gain at 1–6 years; obesity; hyperphagia | 1 | Sleep apnoea | 0.5 |
| Characteristic dysmorphic facial features | 1 | Short stature | 0 |
| Hypogonadism with small genitalia, pubertal delay and insufficiency | 1 | Hypopigmentation | 0 |
| Small hands and feet | 0.5 | ||
| Developmental delay/intellectual disability | 1 | Narrow hands, straight ulnar border | 0.5 |
| Deletion or other cytogenetic/molecular abnormality of the Prader-Willi chromosome region, including maternal disomy | 0 | Esotropia, myopia | 0.5 |
| Thick, viscous saliva | 0 | ||
| Speech articulation defects | 0.5 | ||
| Skin picking | 0.5 | ||
| Total major | 6 | Total minor | 4 |
| Total points index case | 10 points | ||
| Requirements clinical diagnosis PWS for adults | 8 points, of which 5 points major | ||
| Clinical Features [1,2,3,5,6,39,40,41] | Present in Index Case | Prevalent in PWS |
|---|---|---|
| Poor foetal movement | yes | yes |
| Hypotonia during infancy | yes | yes |
| Feeding problems during infancy | yes | yes |
| Abnormal pubertal development | yes | yes |
| Developmental delay Speech Psychomotor | yes yes | yes yes |
| Intellectual disability | yes | yes |
| Sleep related breathing disorder | yes | yes |
| Osteoporosis | yes | yes |
| Diabetes mellitus type 2 | yes | yes |
| Hypertension | no | yes |
| Abnormal pain registration | no | yes |
| Pituitary hormone deficiencies Hypogonadism Growth hormone deficiency Hypothyroidism | yes yes no | yes yes yes |
| Vitamin D deficiency | yes | yes |
| Bowel problems Obstipation | yes | yes |
| Abnormal temperature regulation | no | yes |
| Leg edema | no | yes |
| Foot problems | yes | yes |
| Challenging behaviour Skin picking Hair pulling Temper tantrums Stealing food | yes yes yes yes | yes yes yes yes |
| Obesity | yes | yes |
| Hyperphagia | yes | yes |
| Dysmorphic features Narrow temple distance Narrow nasal bridge Almond-shaped eyes Small palpebral fissure length Strabismus Thin upper lip Low hair line Small chin Broad nose Small hands and feet Scoliosis Kyphosis | yes no yes yes yes no yes yes yes yes no yes | yes yes yes no yes yes no no no yes yes yes |
| Short stature (height below −2 SD) | no | yes |
| Case and Reference | Sensitivity [46] | Greger et al. (1993) & Hamabe et al. (1991) [47,48] | Schulze et al. (1996) [49] | Sun et al. (1996) [50] | Conroy et al. (1997) [51] | Kuslich et al. (1999) [52] | Wirth et al. (2001) [53] |
|---|---|---|---|---|---|---|---|
| Genetic abnormality | 1.5 Mb deletion paternally transmitted | Balanced translocation t(9;15)(q21;q12–13) | Balanced translocation t(15;19)(q12;q13.41) | Balanced translocation t(2;15)(q37.2;q11.2) | Balanced translocation t(4;15)(q27;q11.2) | Balanced translocation t(X;15)(q28;q12) | |
| Location | SNORD115 to GABRB3 intron 3 | SNRPN exon 20/intron 20 (located between SNORD108 and SNORD109A) | SNRPN intron 2 | SNRPN exon 20/intron 20 (located between SNORD108 and SNORD109A) | SNRPN intron 2 | SNRPN exon 20/intron 20 (located between SNORD108 and SNORD109A) | |
| Gender | 3 family members | male | male | male | male | female | |
| Age at last examination (years) | 29 | 3 | 4 | 11 | 20 | ||
| Weight (kg) | 89 | 34 | >95th percentile | NA | 72 | ||
| Height (cm) | 169 | 103 | 50–75th percentile | NA | 151 | ||
| Major criteria Neonatal hypotonia Feeding problems/FTT Excessive weight gain 1–6 years Characteristic facial features Hypogonadism/genital hypoplasia/delayed or incomplete puberty Developmental delay/intellectual disability/learning problems Hyperphagia/food foraging/obsession with food | 98 96 95 49 96 98 93 | no phenotype when paternally transmitted (causes Angelman syndrome when maternally transmitted) | + + - (at 7 years) + + + NA | + + + + + + + | + + + + NA + + | + + + + + + + | - - + - + + + |
| Minor criteria Decreased foetal movements/infantile lethargy Challenging behaviour Sleep disturbance or sleep apnoea Short stature Hypopigmentation Small hands/feet for height and age Narrow hand, straight ulnar border Eye abnormalities (esotropia, myopia) Thick, viscous saliva Speech articulation defects Skin picking | 89 82 37 86 47 75 69 49 83 93 61 | NA + + + + + NA + NA - + | + + - - - + NA - - - - | + + NA − NA − NA + + + + | + + + NA − − − − + + + | − + − + − − NA + NA − − | |
| Case and Reference | Maina et al. (2007) id05 and Whittington et al. (2002) Case 1 [54,55] | Bürger et al. (2002) & Runte et al. (2005) [56,57] | Schüle et al. (2005) [58] | Sahoo et al. (2008) [14] | Calounova et al. (2008) patient 2 [59] | de Smith et al. (2009) [15] | |
| Genetic abnormality | Deletion of maximum 1.8 Mb | ~570 kb deletion paternally transmitted | Balanced translocation t(4;15)(q27;11.2) | 175 kb deletion | 9.5 Mb deletion | 187 kb deletion | |
| Location | Includes: CYFIP1 and MKRN3, but not SNRPN | SNORD115 UTAI UBE3A | SNRPN intron 17 (=between SNORD108 and SNORD109A) | SNORD109A UTAI SNORD115-24 | NPAP1 UTAI AVEN | SNURF-SNRPN exon 2 UTAI IPW | |
| Gender | male | 3 family members | male | male | female | male | |
| Age at last examination (years) | 22 | 22 | 4 | 18 | 19 | ||
| Weight (kg) | NA | 90 | 63 | 79 | 109 | ||
| Height (cm) | NA | 164 | 115 | 149 | 167.5 | ||
| Major criteria Neonatal hypotonia Feeding problems/FTT Excessive weight gain 1–6 years Characteristic facial features Hypogonadism/genital hypoplasia/delayed or incomplete puberty Developmental delay/intellectual disability/ learning problems Hyperphagia/food foraging/obsession with food | + + + NA + + + | no phenotype when paternally transmitted (causes Angelman syndrome when maternally transmitted) | + + −(at 8 years) − + +/− + | + + + + + + + | + + NA NA + + NA | + + + +/− + + + | |
| Minor criteria Decreased foetal movements/infantile lethargy Challenging behaviour Sleep disturbance or sleep apnoea Short stature Hypopigmentation Small hands/feet for height and age Narrow hand, straight ulnar border Eye abnormalities (esotropia, myopia) Thick, viscous saliva Speech articulation defects Skin picking | + + + + + + NA + + + + | + + + + − + − + − − + | + + + − NA + NA − − + + | NA + NA + NA NA NA + NA NA NA | NA + NA − NA + NA NA NA NA + | ||
| Case and Reference | Patient 1 in Kanber et al. (2009) [60] | Duker et al. (2010) [16] | Naik et al. (2011) [61] | Rossi et al. (2012) & Rivera et al. (1990) & Fraccaro et al. (1983) [45,62,63] | Buiting et al. (2014) [64] | Bieth et al. (2015) [17] | |
| Genetic abnormality | Unbalanced translocation t(X;15)(q28;q11.2) | 236 kb deletion | 25 bp duplication | Jumping translocation with a major t(15;18)(q13;q23) and a minor t(X;15)(q28;q13) cell line | ~3.9 Mb deletion | 118 kb deletion | |
| Location | Breakpoint: between NDN and SNURF-SNRPN Deletion: CYFIP1 UTAI NDN | 3′ end of SNRPN (exon 10) UTAI SNORD155-24 | Exon 1 SNURF/SNRPN | Between SNORD108 & SNORD116 | CHEK2P2 UTAI SNRPN exon U1B* | SNORD109A UTAI IPW | |
| Gender | female | male | female | female | male | female | |
| Age at last examination (years) | 12 | 11 | 11 | 10 months | 3 | 23 | |
| Weight (kg) | 75 | 94 | 63.5 | 9.9 | 17.3 | 75 | |
| Height (cm) | 160 | 10–25th percentile | 170 | 70 | 103 | 155 (6m GHT) | |
| Major criteria Neonatal hypotonia Feeding problems/FTT Excessive weight gain 1–6 years Characteristic facial features Hypogonadism/genital hypoplasia/delayed or incomplete puberty Developmental delay/intellectual disability/learning problems Hyperphagia/food foraging/obsession with food | − + NA − − + − | + + −(7 months) − + + + | − − + − NA + +/− | + + NA + + + NA | Asymptomatic apart from delayed motor skills and transient muscular hypotonia associated with mild feeding difficulties in infancy | + + + + + + + | |
| Minor criteria Decreased foetal movements/infantile lethargy Challenging behaviour Sleep disturbance or sleep apnoea Short stature Hypopigmentation Small hands/feet for height and age Narrow hand, straight ulnar border Eye abnormalities (esotropia, myopia) Thick, viscous saliva Speech articulation defects Skin picking | − − − − − − NA NA − − − | + + + NA − − NA + + + − | − + NA − NA NA NA NA NA NA NA | + NA NA NA + NA NA NA NA NA NA | NA + + + NA + NA + + NA + | ||
| Case and Reference | Koufaris et al. (2016) [65] | Fontana et al. (2017) [19] | Lei et al. (2019) [66] | Tan et al. (2020) [13] | |||
| Genetic abnormality | 13 kb deletion | ~ 81 kb deletion | Balanced translocation t(15;19)(q11.2;q13.3) | 71 kb deletion | |||
| Location | U1B and U1B* upstream exons of SNRPN | SNORD109A to exon 3 IPW | Between PAR5 (including SNORD108) and PAR6 | At least SNORD116 and IPW | |||
| Gender | NA | male | male | male | |||
| Age at last examination (years) | child | 18 | 13 | 17 | |||
| Weight (kg) | overweight | 76 | 45 | BMI: 28.45 kg/m2 | |||
| Height (cm) | NA | 174 | 132 | 181 | |||
| Major criteria Neonatal hypotonia Feeding problems/FTT Excessive weight gain 1-6 years Characteristic facial features Hypogonadism/genital hypoplasia/delayed or incomplete puberty Developmental delay/intellectual disability/ learning problems Hyperphagia/food foraging/obsession with food | Overweight child with mild intellectual disability and neurodevelopmental delay | + NA + − + +/− + | + + − (7 months) NA + + + | + + + + − + + | |||
| Minor criteria Decreased foetal movements/infantile lethargy Challenging behaviour Sleep disturbance or sleep apnoea Short stature Hypopigmentation Small hands/feet for height and age Narrow hand, straight ulnar border Eye abnormalities (esotropia, myopia) Thick, viscous saliva Speech articulation defects Skin picking | | NA NA NA − NA − NA + NA NA NA | + + − + − − − − − − − | + + + − NA − NA − NA NA + | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellikaan, K.; van Woerden, G.M.; Kleinendorst, L.; Rosenberg, A.G.W.; Horsthemke, B.; Grosser, C.; van Zutven, L.J.C.M.; van Rossum, E.F.C.; van der Lely, A.J.; Resnick, J.L.; et al. The Diagnostic Journey of a Patient with Prader–Willi-Like Syndrome and a Unique Homozygous SNURF-SNRPN Variant; Bio-Molecular Analysis and Review of the Literature. Genes 2021, 12, 875. https://doi.org/10.3390/genes12060875
Pellikaan K, van Woerden GM, Kleinendorst L, Rosenberg AGW, Horsthemke B, Grosser C, van Zutven LJCM, van Rossum EFC, van der Lely AJ, Resnick JL, et al. The Diagnostic Journey of a Patient with Prader–Willi-Like Syndrome and a Unique Homozygous SNURF-SNRPN Variant; Bio-Molecular Analysis and Review of the Literature. Genes. 2021; 12(6):875. https://doi.org/10.3390/genes12060875
Chicago/Turabian StylePellikaan, Karlijn, Geeske M. van Woerden, Lotte Kleinendorst, Anna G. W. Rosenberg, Bernhard Horsthemke, Christian Grosser, Laura J. C. M. van Zutven, Elisabeth F. C. van Rossum, Aart J. van der Lely, James L. Resnick, and et al. 2021. "The Diagnostic Journey of a Patient with Prader–Willi-Like Syndrome and a Unique Homozygous SNURF-SNRPN Variant; Bio-Molecular Analysis and Review of the Literature" Genes 12, no. 6: 875. https://doi.org/10.3390/genes12060875
APA StylePellikaan, K., van Woerden, G. M., Kleinendorst, L., Rosenberg, A. G. W., Horsthemke, B., Grosser, C., van Zutven, L. J. C. M., van Rossum, E. F. C., van der Lely, A. J., Resnick, J. L., Brüggenwirth, H. T., van Haelst, M. M., & de Graaff, L. C. G. (2021). The Diagnostic Journey of a Patient with Prader–Willi-Like Syndrome and a Unique Homozygous SNURF-SNRPN Variant; Bio-Molecular Analysis and Review of the Literature. Genes, 12(6), 875. https://doi.org/10.3390/genes12060875