
Weighted Gene Co-Expression Network Analysis Reveals Hub Genes Contributing to Fuzz Development in Gossypium arboreum

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction and RNA-Seq
2.3. RNA-Seq Data Analysis
2.4. qRT-PCR
2.5. Construction of Co-Expression Network and Network Analyses
3. Results
3.1. Data Processing and Analysis
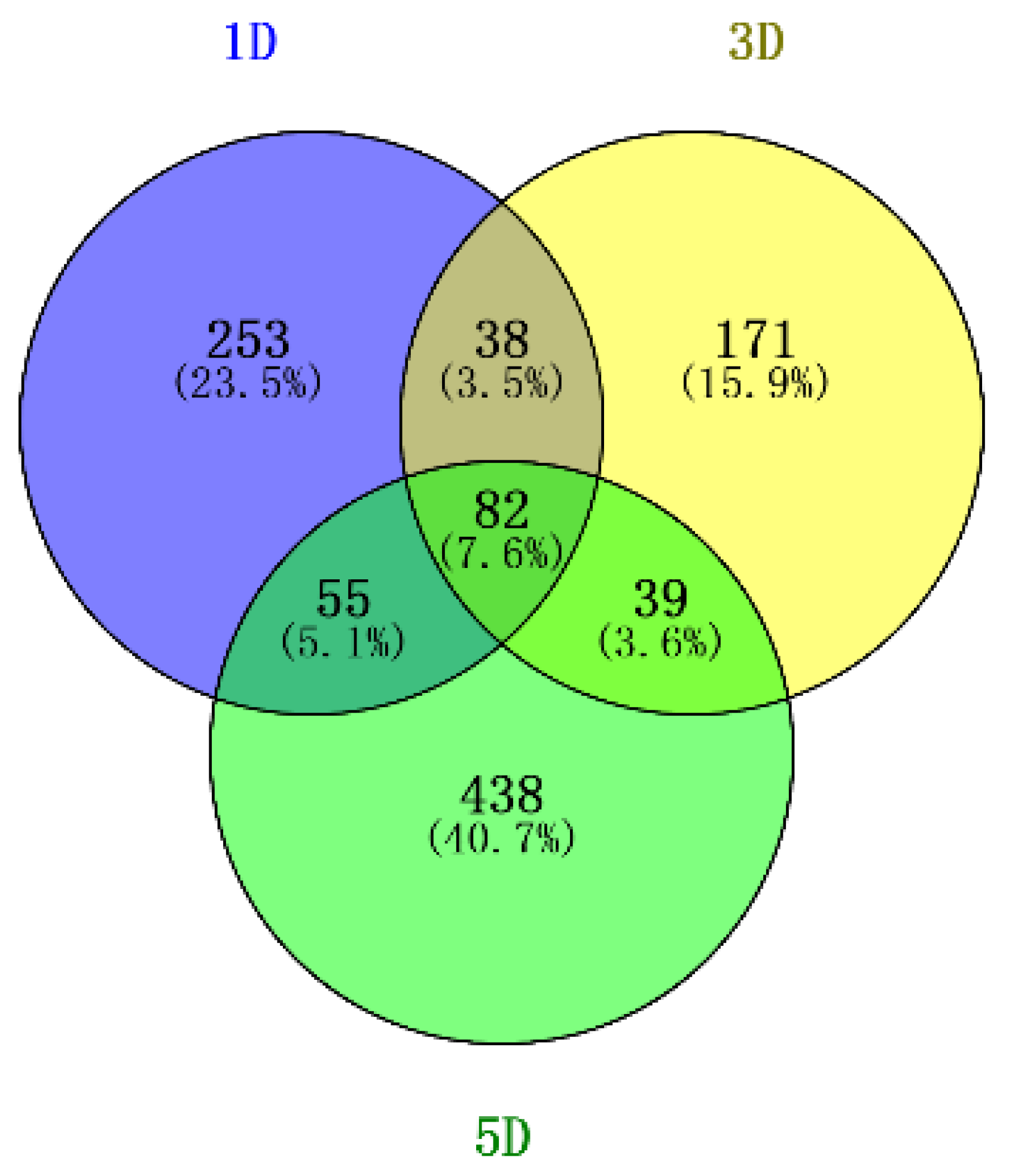
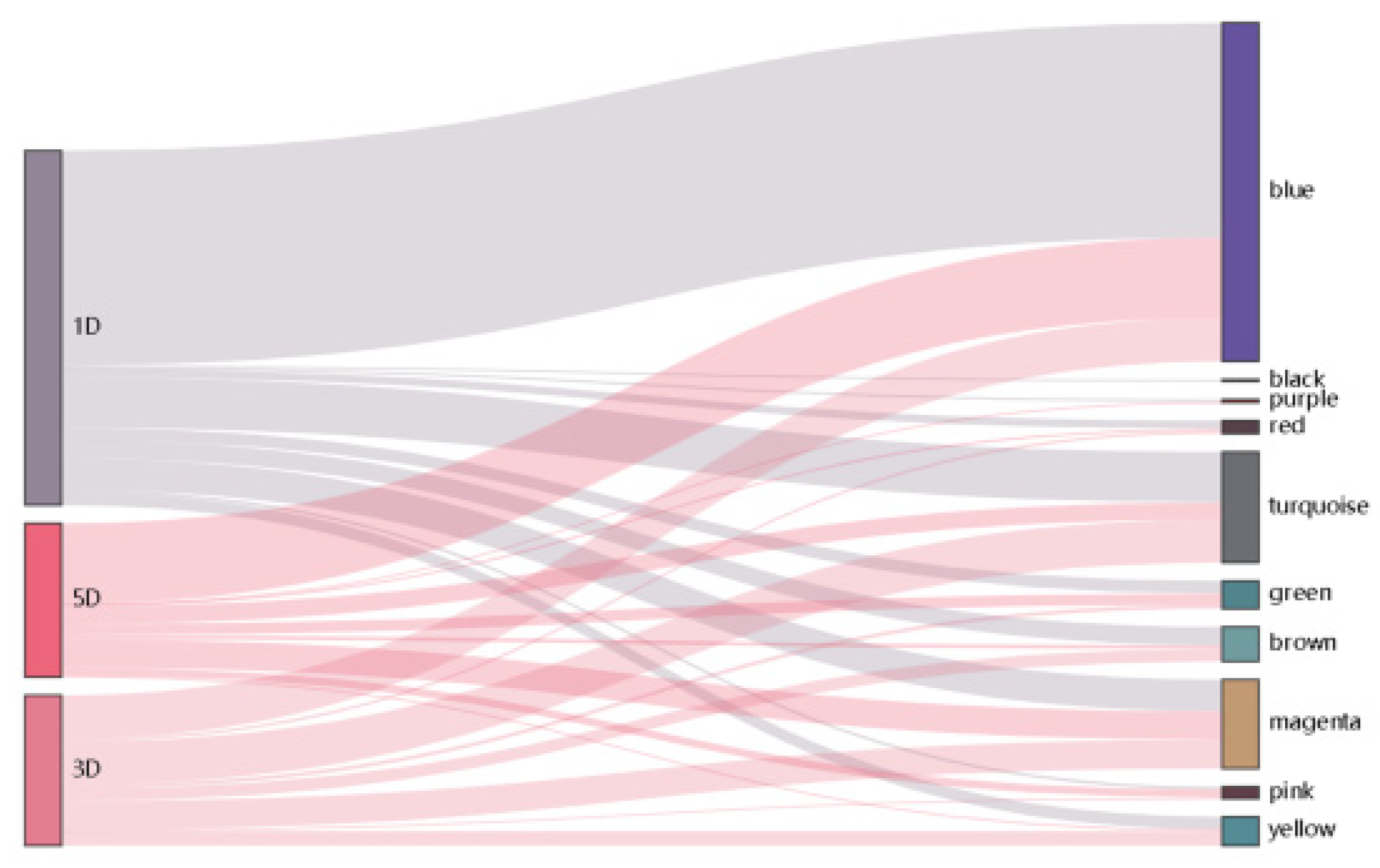
3.2. Differential Gene Expression in DPL971 and DPL972 during Fuzz Initiation
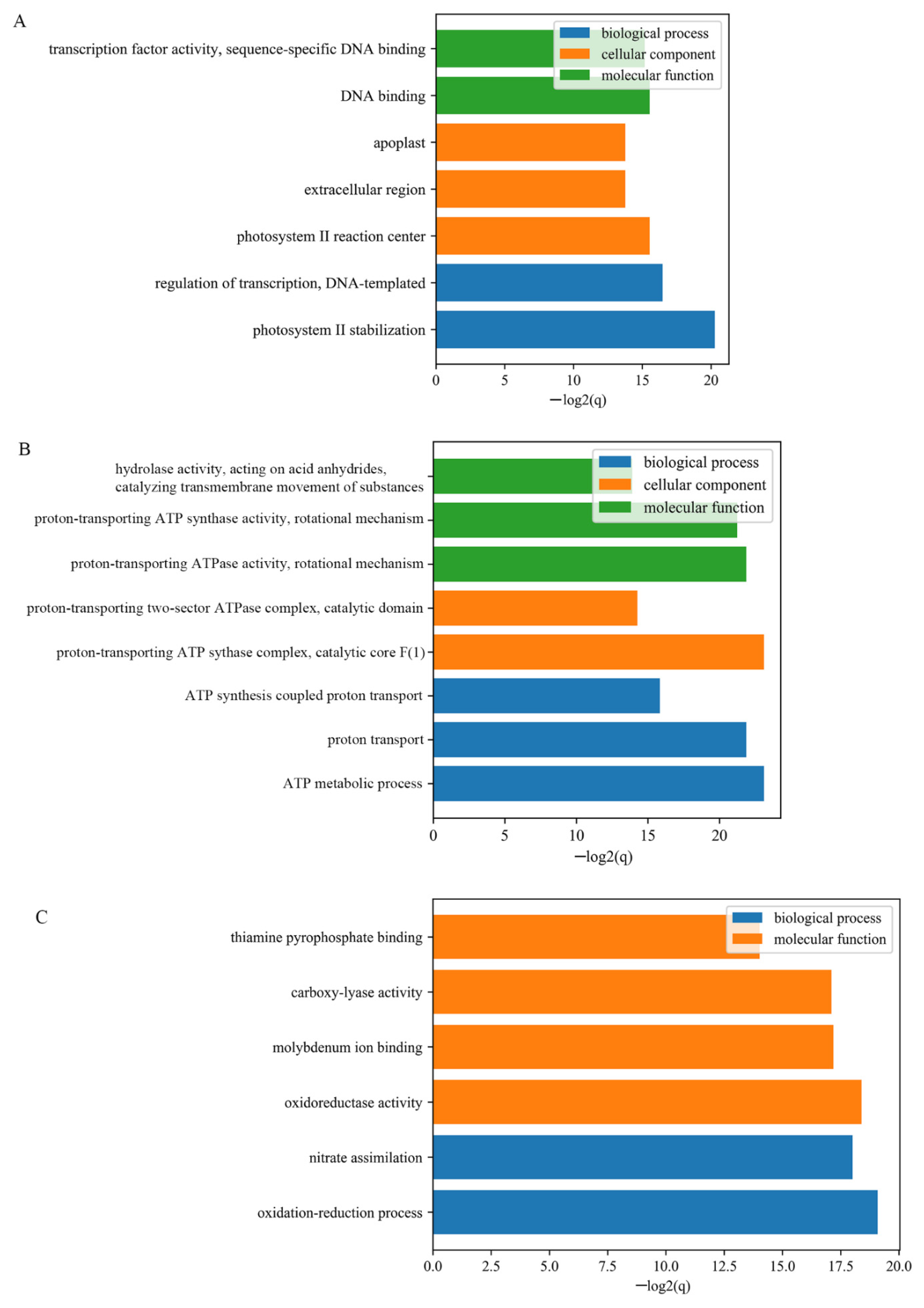
3.3. Functional Enrichment of DEGs
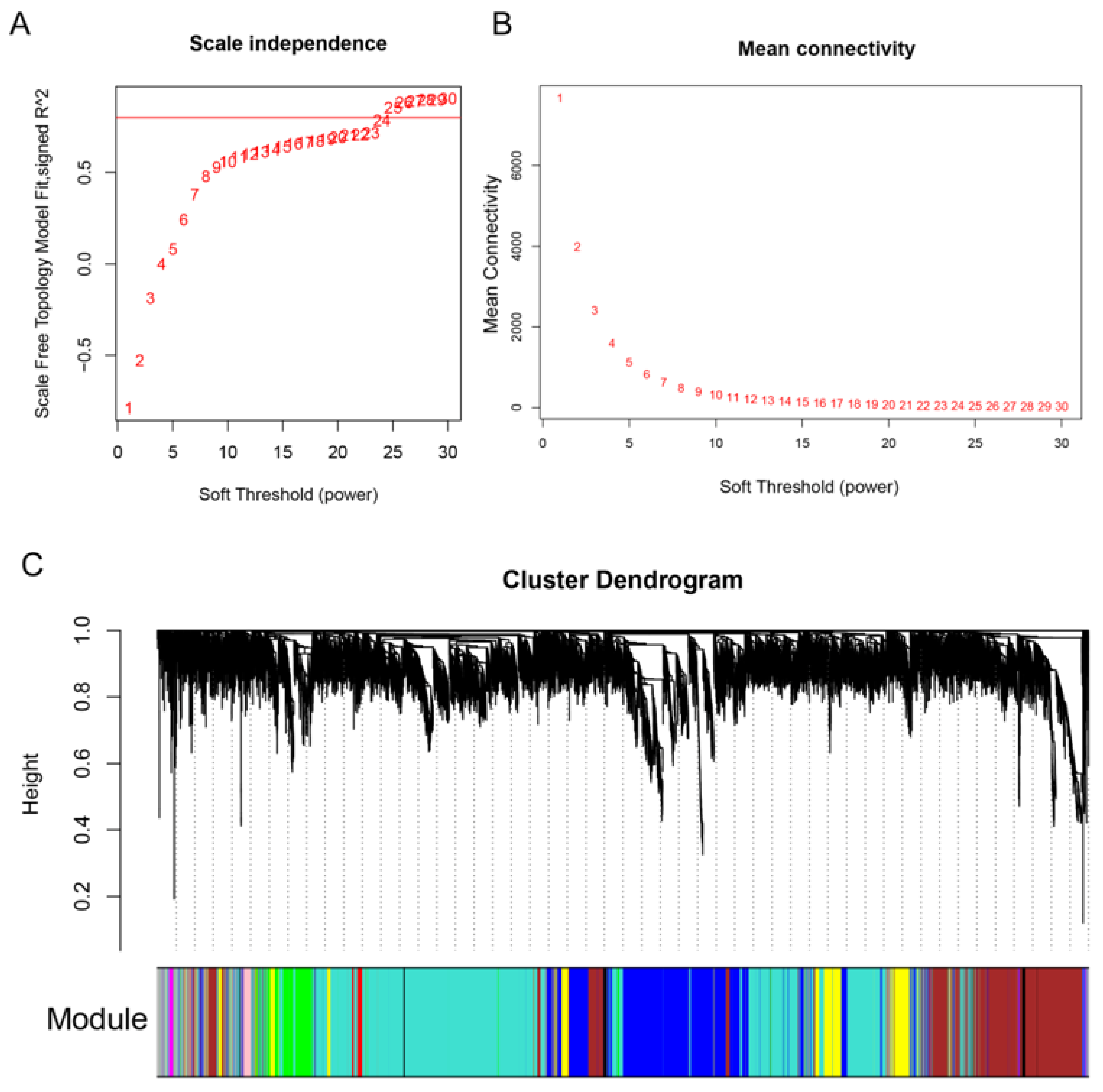
3.4. Co-Expression Network Construction and Module Mining
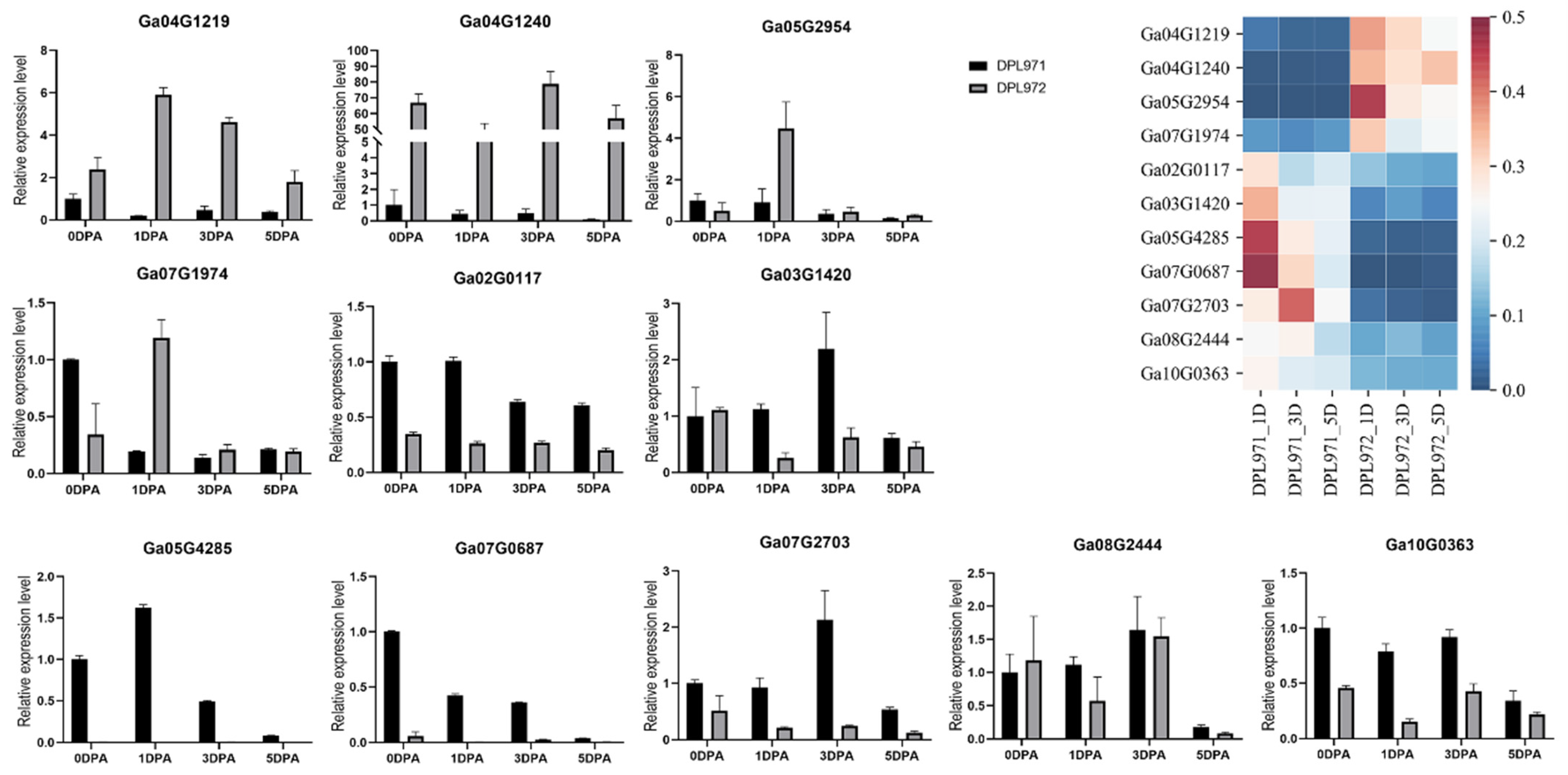
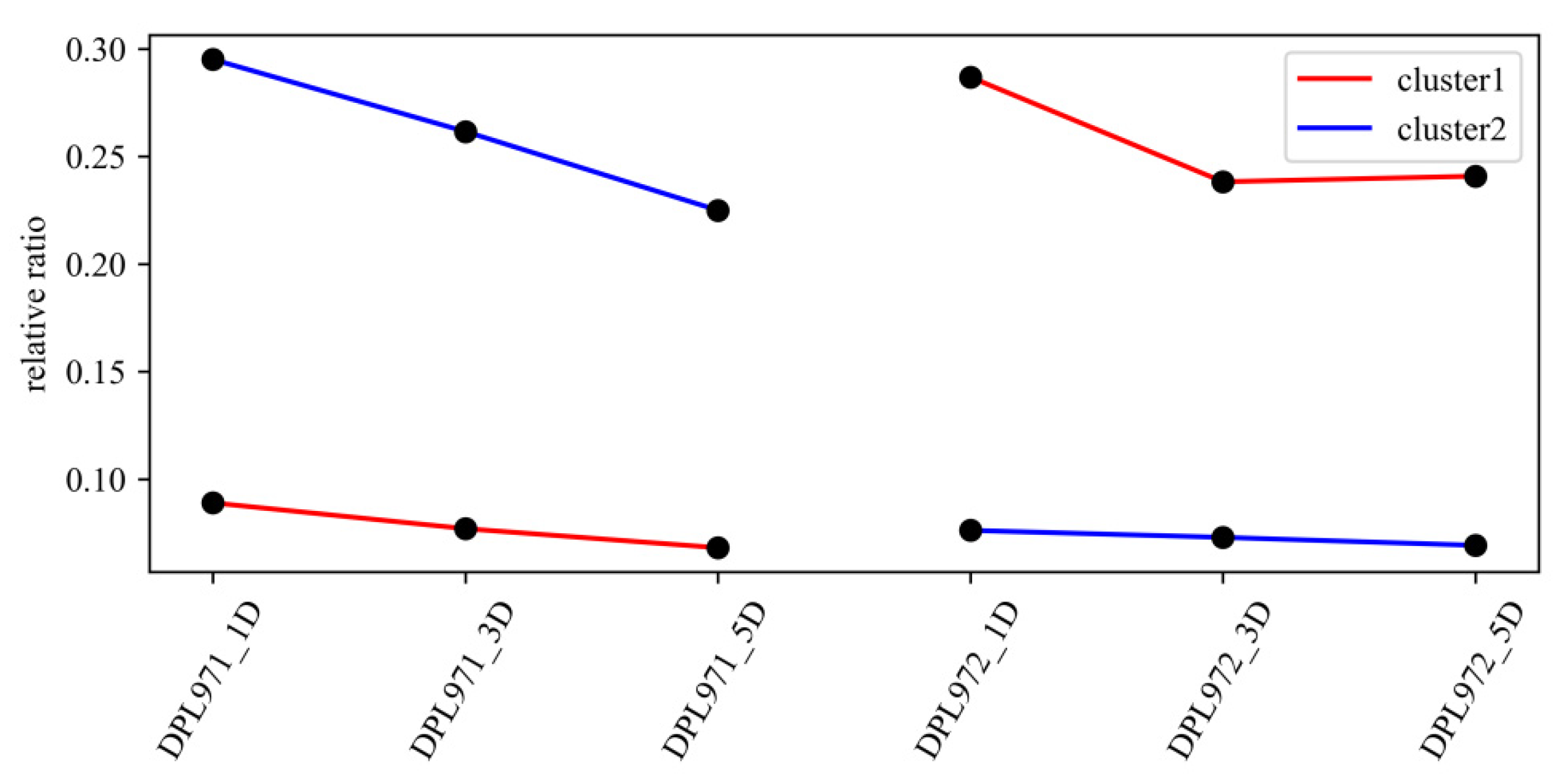
3.5. Functional Enrichment and Expression Analysis of Fuzz-Associated Hub Genes
4. Discussion
4.1. Hub Genes in MEgrey60 Cluster Might Be Involved in Regulatory Network Responsible to Fuzz Initiation
4.2. Multiple Protein Kinase Genes may Regulate Fuzz and Plant Development in G. arboreum
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dai, X.; Zhou, L.; Zhang, W.; Cai, L.; Guo, H.; Tian, H.; Schiefelbein, J.; Wang, S. A single amino acid substitution in the R3 domain of GLABRA1 leads to inhibition of trichome formation in Arabidopsis without affecting its interaction with GLABRA3. Plant Cell Environ. 2015, 39, 897–907. [Google Scholar] [CrossRef]
- Tan, J.; Walford, S.A.; Dennis, E.S.; Llewellyn, D.J. Trichomes at the Base of the Petal Are Regulated by the Same Transcription Factors as Cotton Seed Fibers. Plant Cell Physiol. 2020, 61, 1590–1599. [Google Scholar] [CrossRef]
- Matias-Hernandez, L.; Aguilar-Jaramillo, A.E.; Cigliano, R.A.; Sanseverino, W.; Pelaz, S. Flowering and trichome development share hormonal and transcription factor regulation. J. Exp. Bot. 2016, 67, 1209–1219. [Google Scholar] [CrossRef]
- Yang, C.; Ye, Z. Trichomes as models for studying plant cell differentiation. Cell Mol. Life Sci. 2013, 70, 1937–1948. [Google Scholar] [CrossRef]
- Hulskamp, M. Plant trichomes: A model for cell differentiation. Nat. Rev. Mol. Cell Biol. 2004, 5, 471–480. [Google Scholar] [CrossRef]
- Balkunde, R.; Pesch, M.; Hülskamp, M. Chapter Ten-Trichome Patterning in Arabidopsis thaliana: From Genetic to Molecular Models. In Current Topics in Developmental Biology; Marja, C.P.T., Ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 91, pp. 299–321. [Google Scholar]
- Pattanaik, S.; Patra, B.; Singh, S.K.; Yuan, L. An overview of the gene regulatory network controlling trichome development in the model plant, Arabidopsis. Front. Plant Sci. 2014, 5, 259. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Sano, R.; Wada, T.; Takabayashi, J.; Okada, K. Jasmonic acid control of GLABRA3 links inducible defense and trichome patterning in Arabidopsis. Development 2009, 136, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Gao, D.; Xiong, Y.; Tang, X.; Xiao, X.; Wang, C.; Yu, S. Hairy Leaf 6, an AP2/ERF transcription factor, interacts with OsWOX3B and regulates trichome formation in rice. Mol. Plant 2017, 10, 1417–1433. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, K.; Wang, Y.; Peng, Y.; Hu, F.; Wen, L.; Han, B.; Qian, Q.; Teng, S. A WUSCHEL-like homeobox gene, OsWOX3B responses to NUDA/GL-1 locus in rice. Rice J. 2012, 5, 30. [Google Scholar] [CrossRef]
- Vernoud, V.; Laigle, G.; Rozier, F.; Meeley, R.B.; Perez, P.; Rogowsky, P.M. The HD-ZIP IV transcription factor OCL4 is necessary for trichome patterning and anther development in maize. Plant J. Cell Mol. Biol. 2009, 59, 883–894. [Google Scholar] [CrossRef]
- Kong, D.; Pan, X.; Jing, Y.; Zhao, Y.; Duan, Y.; Yang, J.; Wang, B.; Liu, Y.; Shen, R.; Cao, Y.; et al. ZmSPL10/14/26 are required for epidermal hair cell fate specification on maize leaf. New Phytol. 2021, 230, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; van Herwijnen, Z.O.; Drager, D.B.; Sui, C.; Haring, M.A.; Schuurink, R.C. SlMYC1 Regulates Type VI Glandular Trichome Formation and Terpene Biosynthesis in Tomato Glandular Cells. Plant Cell 2018, 30, 2988–3005. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, H.; Zhang, J.; Luo, Z.; Gong, P.; Zhang, C.; Li, J.; Wang, T.; Zhang, Y.; Lu, Y.; et al. A regulatory gene induces trichome formation and embryo lethality in tomato. Proc. Natl. Acad. Sci. USA 2011, 108, 11836–11841. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Chen, M.; Shen, Q.; Li, L.; Fu, X.; Pan, Q.; Tang, Y.; Shi, P.; Lv, Z.; Jiang, W.; et al. HOMEODOMAIN PROTEIN 1 is required for jasmonate-mediated glandular trichome initiation in Artemisia annua. New Phytol. 2017, 213, 1145–1155. [Google Scholar] [CrossRef]
- Wang, L.K.D.; Ruan, Y. Looking into ‘hair tonics’ for cotton fiber initiation. New Phytol. 2020, 229, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wu, S.; Nowak, J.; Wang, G.; Han, L.; Feng, Z.; Mendrinna, A.; Ma, Y.; Wang, H.; Zhang, X.; et al. Live-cell imaging of the cytoskeleton in elongating cotton fibres. Nat. Plants 2019, 5, 498–504. [Google Scholar] [CrossRef]
- Wang, L.; Wang, G.; Long, L.; Altunok, S.; Feng, Z.; Wang, D.; Khawar, K.M.; Mujtaba, M. Understanding the role of phytohormones in cotton fiber development through omic approaches; recent advances and future directions. Int. J. Biol. Macromol. 2020, 163, 1301–1313. [Google Scholar] [CrossRef]
- Mei, G.; Zhang, Z. Optimization of polar distribution of GhPIN3a in the ovule epidermis improves cotton fiber development. J. Exp. Bot. 2019, 70, 3021–3023. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, M.; Hou, L.; Bai, W.; Yan, X.; Hou, N.; Wang, H.; Huang, J.; Zhao, J.; Pei, Y. Cytokinin inhibits cotton fiber initiation by disrupting PIN3a-mediated asymmetric accumulation of auxin in the ovule epidermis. J. Exp. Bot. 2019, 70, 3139–3151. [Google Scholar] [CrossRef]
- Xiao, G.; He, P.; Zhao, P.; Liu, H.; Zhang, L.; Pang, C.; Yu, J. Genome-wide identification of the GhARF gene family reveals that GhARF2 and GhARF18 are involved in cotton fibre cell initiation. J. Exp. Bot. 2018, 69, 4323–4337. [Google Scholar] [CrossRef]
- Zhang, M.; Zeng, J.Y.; Long, H.; Xiao, Y.H.; Yan, X.Y.; Pei, Y. Auxin Regulates Cotton Fiber Initiation via GhPIN-Mediated Auxin Transport. Plant Cell Physiol. 2017, 58, 385–397. [Google Scholar] [CrossRef]
- Walford, S.A.; Wu, Y.; Llewellyn, D.J.; Dennis, E.S. Epidermal cell differentiation in cotton mediated by the homeodomain leucine zipper gene, GhHD-1. Plant J. Cell Mol. Biol. 2012, 71, 464–478. [Google Scholar] [CrossRef]
- Shangguan, X.X.; Yang, C.Q.; Zhang, X.F.; Wang, L.J. Functional characterization of a basic helix-loop-helix (bHLH) transcription factor GhDEL65 from cotton (Gossypium hirsutum). Physiol. Plant 2016, 158, 200–212. [Google Scholar] [CrossRef]
- Sun, H.; Hao, P.; Gu, L.; Cheng, S.; Wang, H.; Wu, A.; Ma, L.; Wei, H.; Yu, S. Pectate lyase-like Gene GhPEL76 regulates organ elongation in Arabidopsis and fiber elongation in cotton. Plant Sci. Int. J. Exp. Plant Biol. 2020, 293, 110395. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, H.; Xiong, F.; Ma, S.; Zheng, L.; Song, Y.; Deng, K.; Wu, H.; Li, F.; Yang, Z. Comparative phosphoproteomic analysis of BR-defective mutant reveals a key role of GhSK13 in regulating cotton fiber development. Sci. China Life Sci. 2020, 63, 1905–1917. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Z.; Xiao, Y.; Wang, P.; Wang, Y.; Ge, X.; Zhang, C.; Zhang, X.; Li, F. Genome-Wide Analysis of the NF-YB Gene Family in Gossypium hirsutum L. and Characterization of the Role of GhDNF-YB22 in Embryogenesis. Int. J. Mol. Sci. 2018, 19, 483. [Google Scholar] [CrossRef]
- Hu, W.; Chen, L.; Qiu, X.; Wei, J.; Lu, H.; Sun, G.; Ma, X.; Yang, Z.; Zhu, C.; Hou, Y.; et al. AKR2A participates in the regulation of cotton fibre development by modulating biosynthesis of very-long-chain fatty acids. Plant Biotechnol. J. 2020, 18, 526–539. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, G.-Q.; Zou, D.; Yan, J.-Q.; Li, Y.; Hu, S.; Li, X.-B. The cotton (Gossypium hirsutum) NAC transcription factor (FSN1) as a positive regulator participates in controlling secondary cell wall biosynthesis and modification of fibers. New Phytol. 2017, 217, 625–640. [Google Scholar] [CrossRef]
- Shan, C.-M.; Shangguan, X.-X.; Zhao, B.; Zhang, X.-F.; Chao, L.-m.; Yang, C.-Q.; Wang, L.-J.; Zhu, H.-Y.; Zeng, Y.-D.; Guo, W.-Z.; et al. Control of cotton fibre elongation by a homeodomain transcription factor GhHOX3. Nat. Commun. 2014, 5, 5519. [Google Scholar] [CrossRef]
- Cao, J.F.; Zhao, B.; Huang, C.C.; Chen, Z.W.; Zhao, T.; Liu, H.R.; Hu, G.J.; Shangguan, X.X.; Shan, C.M.; Wang, L.J.; et al. The miR319-Targeted GhTCP4 Promotes the Transition from Cell Elongation to Wall Thickening in Cotton Fiber. Mol. Plant 2020, 13, 1063–1077. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, T. MIXTAs and phytohormones orchestrate cotton fiber development. Curr. Opin. Plant Biol. 2021, 59, 101975. [Google Scholar] [CrossRef]
- Wan, Q.; Guan, X.; Yang, N.; Wu, H.; Pan, M.; Liu, B.; Fang, L.; Yang, S.; Hu, Y.; Ye, W.; et al. Small interfering RNAs from bidirectional transcripts of GhMML3_A12 regulate cotton fiber development. New Phytol. 2016, 210, 1298–1310. [Google Scholar] [CrossRef]
- Chen, W.; Li, Y.; Zhu, S.; Fang, S.; Zhao, L.; Guo, Y.; Wang, J.; Yuan, L.; Lu, Y.; Liu, F.; et al. A Retrotransposon Insertion in GhMML3_D12 Is Likely Responsible for the Lintless Locus li3 of Tetraploid Cotton. Front. Plant Sci. 2020, 11, 593679. [Google Scholar] [CrossRef]
- Wu, H.; Tian, Y.; Wan, Q.; Fang, L.; Guan, X.; Chen, J.; Hu, Y.; Ye, W.; Zhang, H.; Guo, W.; et al. Genetics and evolution of MIXTA genes regulating cotton lint fiber development. New Phytol. 2017, 217, 883–895. [Google Scholar] [CrossRef]
- Zhu, Q.-H.; Yuan, Y.; Stiller, W.; Jia, Y.; Wang, P.; Pan, Z.; Du, X.; Llewellyn, D.; Wilson, I. Genetic dissection of the fuzzless seed trait in Gossypium barbadense. J. Exp. Bot. 2018, 69, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D.; Li, P.; Florane, C.B. Elucidation of sequence polymorphism in fuzzless-seed cotton lines. Mol. Genet. Genom. MGG 2021, 296, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Cao, Y.; He, S.; Sun, J.; Dai, H.; Zhang, H.; Sun, C.; Jiang, Y.; Paterson, A.H.; Rong, J. GaHD1, a candidate gene for the Gossypium arboreum SMA-4 mutant, promotes trichome and fiber initiation by cellular H2O2 and Ca(2+) signals. Plant Mol. Biol. 2020, 103, 409–423. [Google Scholar] [CrossRef]
- Liu, X.; Moncuquet, P.; Zhu, Q.H.; Stiller, W.; Zhang, Z.; Wilson, I. Genetic Identification and Transcriptome Analysis of Lintless and Fuzzless Traits in Gossypium arboreum L. Int. J. Mol. Sci. 2020, 21, 1675. [Google Scholar] [CrossRef]
- Feng, X.; Cheng, H.; Zuo, D.; Zhang, Y.; Wang, Q.; Liu, K.; Ashraf, J.; Yang, Q.; Li, S.; Chen, X.; et al. Fine mapping and identification of the fuzzless gene GaFzl in DPL972 (Gossypium arboreum). TAG Theor. Appl. Genet. Theor. Angew. Genet. 2019, 132, 2169–2179. [Google Scholar] [CrossRef]
- Wang, X.; Miao, Y.; Cai, Y.; Sun, G.; Jia, Y.; Song, S.; Pan, Z.; Zhang, Y.; Wang, L.; Fu, G.; et al. Large-fragment insertion activates gene GaFZ (Ga08G0121) and is associated with the fuzz and trichome reduction in cotton (Gossypium arboreum). Plant Biotechnol. J. 2020, 1–15. [Google Scholar] [CrossRef]
- Du, X.; Huang, G.; He, S.; Yang, Z.; Sun, G.; Ma, X.; Li, N.; Zhang, X.; Sun, J.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Kuang, J.F.; Wu, C.J.; Guo, Y.F.; Walther, D.; Shan, W.; Chen, J.Y.; Chen, L.; Lu, W.J. Deciphering transcriptional regulators of banana fruit ripening by regulatory network analysis. Plant Biotechnol. J. 2021, 19, 477–489. [Google Scholar] [CrossRef]
- Zaidi, S.S.; Naqvi, R.Z.; Asif, M.; Strickler, S.; Shakir, S.; Shafiq, M.; Khan, A.M.; Amin, I.; Mishra, B.; Mukhtar, M.S.; et al. Molecular insight into cotton leaf curl geminivirus disease resistance in cultivated cotton (Gossypium hirsutum). Plant Biotechnol. J. 2020, 18, 691–706. [Google Scholar] [CrossRef]
- Huang, G.Q.; Xu, W.L.; Gong, S.Y.; Li, B.; Wang, X.L.; Xu, D.; Li, X.B. Characterization of 19 novel cotton FLA genes and their expression profiling in fiber development and in response to phytohormones and salt stress. Physiol. Plant 2008, 134, 348–359. [Google Scholar] [CrossRef]
- Bowling, A.J.; Vaughn, K.C.; Turley, R.B. Polysaccharide and glycoprotein distribution in the epidermis of cotton ovules during early fiber initiation and growth. Protoplasma 2011, 248, 579–590. [Google Scholar] [CrossRef]
- Huang, G.Q.; Gong, S.Y.; Xu, W.L.; Li, W.; Li, P.; Zhang, C.J.; Li, D.D.; Zheng, Y.; Li, F.G.; Li, X.B. A fasciclin-like arabinogalactan protein, GhFLA1, is involved in fiber initiation and elongation of cotton. Plant Physiol. 2013, 161, 1278–1290. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, K.; Pandey, P.; Rajamani, V.; Padmalatha, K.V.; Dhandapani, G.; Kanakachari, M.; Leelavathi, S.; Kumar, P.A.; Reddy, V.S. Glycoproteome of elongating cotton fiber cells. Mol. Cell Proteom. 2013, 12, 3677–3689. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Shi, R.; Wang, X.; Pan, Y.; Li, Z.; Yang, X.; Zhang, G.; Ma, Z. Characterization and expression analysis of a fiber differentially expressed Fasciclin-like arabinogalactan protein gene in Sea Island cotton fibers. PLoS ONE 2013, 8, e70185. [Google Scholar] [CrossRef]
- Qin, L.X.; Rao, Y.; Li, L.; Huang, J.F.; Xu, W.L.; Li, X.B. Cotton GalT1 encoding a putative glycosyltransferase is involved in regulation of cell wall pectin biosynthesis during plant development. PLoS ONE 2013, 8, e59115. [Google Scholar] [CrossRef] [PubMed]
- Hande, A.S.; Katageri, I.S.; Jadhav, M.P.; Adiger, S.; Gamanagatti, S.; Padmalatha, K.V.; Dhandapani, G.; Kanakachari, M.; Kumar, P.A.; Reddy, V.S. Transcript profiling of genes expressed during fibre development in diploid cotton (Gossypium arboreum L.). BMC Genom. 2017, 18, 675. [Google Scholar] [CrossRef]
- Qin, L.X.; Chen, Y.; Zeng, W.; Li, Y.; Gao, L.; Li, D.D.; Bacic, A.; Xu, W.L.; Li, X.B. The cotton β-galactosyltransferase 1 (GalT1) that galactosylates arabinogalactan proteins participates in controlling fiber development. Plant J. Cell Mol. Biol. 2017, 89, 957–971. [Google Scholar] [CrossRef]
- Yang, C.-Q.; Wu, X.-M.; Ruan, J.-X.; Hu, W.-L.; Mao, Y.-B.; Chen, X.-Y.; Wang, L.-J. Isolation and characterization of terpene synthases in cotton (Gossypium hirsutum). Phytochemistry 2013, 96, 46–56. [Google Scholar] [CrossRef]
- Zou, C.; Lu, C.; Shang, H.; Jing, X.; Cheng, H.; Zhang, Y.; Song, G. Genome-wide analysis of the Sus gene family in cotton. J. Integr. Plant Biol. 2013, 55, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Huang, J.; Qin, L.; Huang, Y.; Zeng, W.; Rao, Y.; Li, J.; Li, X.; Xu, W. Two cotton fiber-associated glycosyltransferases, GhGT43A1 and GhGT43C1, function in hemicellulose glucuronoxylan biosynthesis during plant development. Physiol. Plant 2014, 152, 367–379. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.-Y. Cotton cytosolic pyruvate kinase GhPK6 participates in fast fiber elongation regulation in a ROS-mediated manner. Planta 2016, 244, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Lu, X.K.; Yin, Z.J.; Mu, M.; Zhao, X.J.; Wang, D.L.; Wang, S.; Fan, W.L.; Guo, L.X.; Ye, W.W.; et al. Genome-wide identification and expression analysis of CIPK genes in diploid cottons. Genet. Mol. Res. 2016, 15, 10–4238. [Google Scholar] [CrossRef]
- Cui, Y.; Su, Y.; Wang, J.; Jia, B.; Wu, M.; Pei, W.; Zhang, J.; Yu, J. Genome-Wide Characterization and Analysis of CIPK Gene Family in Two Cultivated Allopolyploid Cotton Species: Sequence Variation, Association with Seed Oil Content, and the Role of GhCIPK6. Int. J. Mol. Sci. 2020, 21, 863. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; He, X.; Wang, X.; Zhang, S.; Guo, X.; Denby, K. ghr-miR5272a-mediated regulation of GhMKK6 gene transcription contributes to the immune response in cotton. J. Exp. Bot. 2017, 68, 5895–5906. [Google Scholar] [CrossRef]
- Chen, L.; Sun, H.; Wang, F.; Yue, D.; Shen, X.; Sun, W.; Zhang, X.; Yang, X. Genome-wide identification of MAPK cascade genes reveals the GhMAP3K14-GhMKK11-GhMPK31 pathway is involved in the drought response in cotton. Plant Mol. Biol. 2020, 103, 211–223. [Google Scholar] [CrossRef]
- Jun, Z.; Zhang, Z.; Gao, Y.; Zhou, L.; Fang, L.; Chen, X.; Ning, Z.; Chen, T.; Guo, W.; Zhang, T. Overexpression of GbRLK, a putative receptor-like kinase gene, improved cotton tolerance to Verticillium wilt. Sci. Rep. 2015, 5, 15048. [Google Scholar] [CrossRef]
- Yuan, N.; Rai, K.M.; Balasubramanian, V.K.; Upadhyay, S.K.; Luo, H.; Mendu, V. Genome-wide identification and characterization of LRR-RLKs reveal functional conservation of the SIF subfamily in cotton (Gossypium hirsutum). BMC Plant Biol. 2018, 18, 185. [Google Scholar] [CrossRef] [PubMed]
- Babilonia, K.; Wang, P.; Liu, Z.; Jamieson, P.; Mormile, B.; Rodrigues, O.; Zhang, L.; Lin, W.; Danmaigona Clement, C.; Menezes de Moura, S.; et al. A nonproteinaceous Fusarium cell wall extract triggers receptor-like protein-dependent immune responses in Arabidopsis and cotton. New Phytol. 2021, 230, 275–289. [Google Scholar] [CrossRef]
- Wang, P.; Zhou, L.; Jamieson, P.; Zhang, L.; Zhao, Z.; Babilonia, K.; Shao, W.; Wu, L.; Mustafa, R.; Amin, I.; et al. The Cotton Wall-Associated Kinase GhWAK7A Mediates Responses to Fungal Wilt Pathogens by Complexing with the Chitin Sensory Receptors. Plant Cell 2020, 32, 3978–4001. [Google Scholar] [CrossRef]
- Feng, H.; Li, C.; Zhou, J.; Yuan, Y.; Feng, Z.; Shi, Y.; Zhao, L.; Zhang, Y.; Wei, F.; Zhu, H. A cotton WAKL protein interacted with a DnaJ protein and was involved in defense against Verticillium dahliae. Int. J. Biol. Macromol. 2021, 167, 633–643. [Google Scholar] [CrossRef]
- Gao, P.; Zhao, P.M.; Wang, J.; Wang, H.Y.; Du, X.M.; Wang, G.L.; Xia, G.X. Co-expression and preferential interaction between two calcineurin B-like proteins and a CBL-interacting protein kinase from cotton. Plant Physiol. Biochem. PPB Soc. Fr. Physiol. Veg. 2008, 46, 935–940. [Google Scholar] [CrossRef]
- Lu, T.; Zhang, G.; Sun, L.; Wang, J.; Hao, F. Genome-wide identification of CBL family and expression analysis of CBLs in response to potassium deficiency in cotton. PeerJ 2017, 5, e3653. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yang, X.; Sun, W.; Miao, Y.; He, L.; Zhang, X. The Calcium Sensor CBL2 and Its Interacting Kinase CIPK6 Are Involved in Plant Sugar Homeostasis via Interacting with Tonoplast Sugar Transporter TST2. Plant Physiol. 2020, 183, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhang, B.; Deng, J.; Chen, L.; Ullah, A.; Yang, X. Genome-wide analysis of CBL and CIPK family genes in cotton: Conserved structures with divergent interactions and expression. Physiol. Mol. Biol. Plants 2021, 27, 359–368. [Google Scholar] [CrossRef]
- Huang, J.; Pang, C.; Fan, S.; Song, M.; Yu, J.; Wei, H.; Ma, Q.; Li, L.; Zhang, C.; Yu, S. Genome-wide analysis of the family 1 glycosyltransferases in cotton. Mol. Genet. Genom. MGG 2015, 290, 1805–1818. [Google Scholar] [CrossRef]
- Lee, J.; Burns, T.H.; Light, G.; Sun, Y.; Fokar, M.; Kasukabe, Y.; Fujisawa, K.; Maekawa, Y.; Allen, R.D. Xyloglucan endotransglycosylase/hydrolase genes in cotton and their role in fiber elongation. Planta 2010, 232, 1191–1205. [Google Scholar] [CrossRef]
- Li, Z.K.; Chen, B.; Li, X.X.; Wang, J.P.; Zhang, Y.; Wang, X.F.; Yan, Y.Y.; Ke, H.F.; Yang, J.; Wu, J.H.; et al. A newly identified cluster of glutathione S-transferase genes provides Verticillium wilt resistance in cotton. Plant J. Cell Mol. Biol. 2019, 98, 213–227. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Wang, X.; Pan, Z.; Geng, X.; Chen, B.; Liu, B.; Du, X.; Song, X. Identification and characterization analysis of sulfotransferases (SOTs) gene family in cotton (Gossypium) and its involvement in fiber development. BMC Plant Biol. 2019, 19, 595. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, X.; Liu, S.; Cheng, H.; Zuo, D.; Zhang, Y.; Wang, Q.; Lv, L.; Song, G. Weighted Gene Co-Expression Network Analysis Reveals Hub Genes Contributing to Fuzz Development in Gossypium arboreum. Genes 2021, 12, 753. https://doi.org/10.3390/genes12050753
Feng X, Liu S, Cheng H, Zuo D, Zhang Y, Wang Q, Lv L, Song G. Weighted Gene Co-Expression Network Analysis Reveals Hub Genes Contributing to Fuzz Development in Gossypium arboreum. Genes. 2021; 12(5):753. https://doi.org/10.3390/genes12050753
Chicago/Turabian StyleFeng, Xiaoxu, Shang Liu, Hailiang Cheng, Dongyun Zuo, Youping Zhang, Qiaolian Wang, Limin Lv, and Guoli Song. 2021. "Weighted Gene Co-Expression Network Analysis Reveals Hub Genes Contributing to Fuzz Development in Gossypium arboreum" Genes 12, no. 5: 753. https://doi.org/10.3390/genes12050753
APA StyleFeng, X., Liu, S., Cheng, H., Zuo, D., Zhang, Y., Wang, Q., Lv, L., & Song, G. (2021). Weighted Gene Co-Expression Network Analysis Reveals Hub Genes Contributing to Fuzz Development in Gossypium arboreum. Genes, 12(5), 753. https://doi.org/10.3390/genes12050753