
Deletion of the Imprinted Phlda2 Gene Increases Placental Passive Permeability in the Mouse

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Genotyping
2.3. Placental Stereology
2.4. Placental Transfer Assays of Radio-Labelled Solutes
2.5. Statistical Analysis
3. Results
3.1. Phlda2−/+ Mutants Show Placental Overgrowth Associated with Transitory Reduction in Fetal Growth
3.2. Placental Overgrowth in Phlda2−/+ Mutants Does Not Affect the Labyrinthine Zone
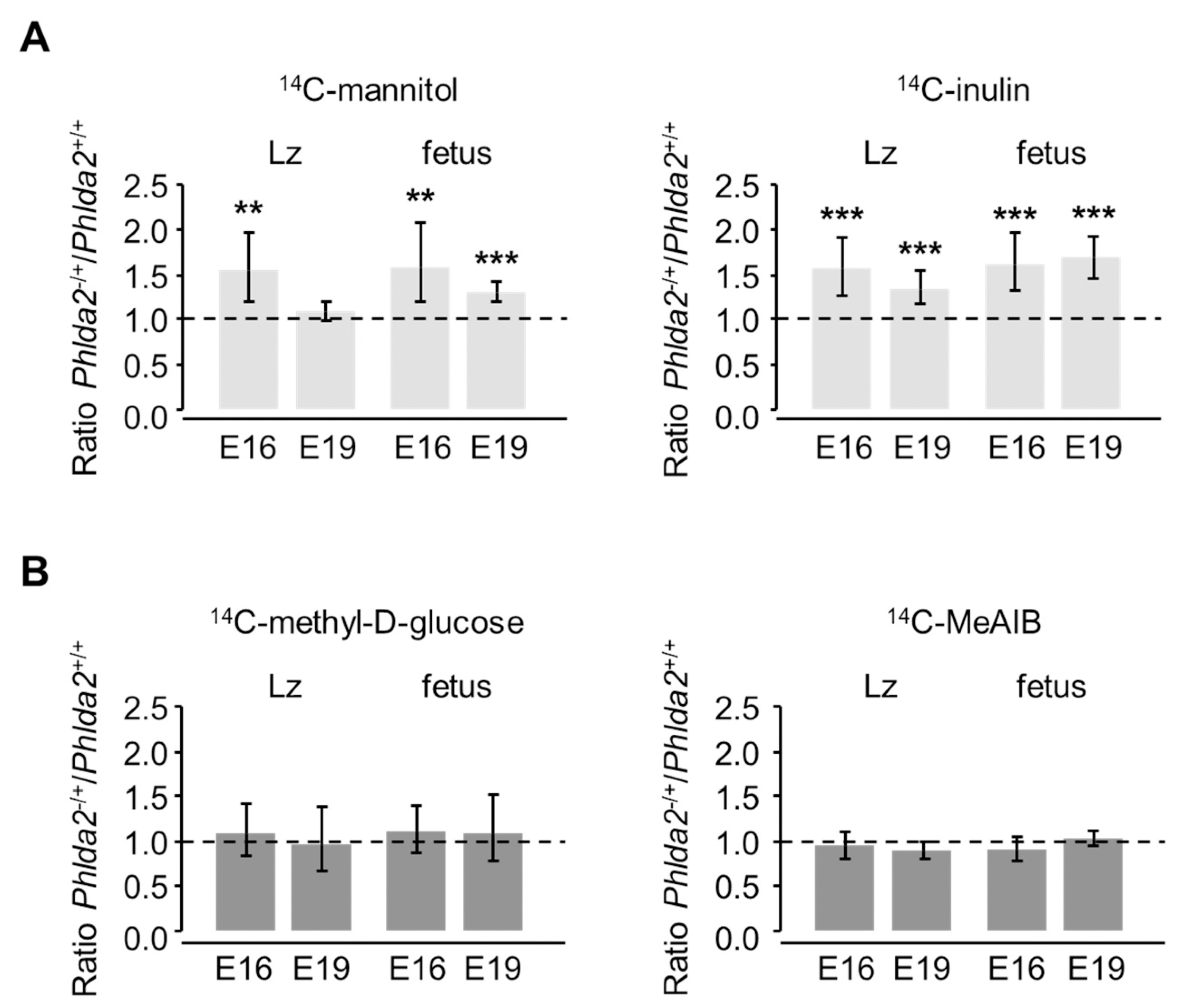
3.3. Phlda2−/+ Placentae Show Increased Passive Permeability to Hydrophilic Solutes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Constância, M.; Kelsey, G.; Reik, W. Resourceful imprinting. Nature 2004, 432, 53–57. [Google Scholar] [CrossRef]
- Gutierrez-Marcos, J.F.; Constância, M.; Burton, G.J. Maternal to offspring resource allocation in plants and mammals. Placenta 2012, 33 (Suppl. 2), e3–e10. [Google Scholar] [CrossRef]
- Babak, T.; DeVeale, B.; Tsang, E.K.; Zhou, Y.; Li, X.; Smith, K.S.; Kukurba, K.R.; Zhang, R.; Li, J.B.; van der Kooy, D.; et al. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat. Genet. 2015, 47, 544–549. [Google Scholar] [CrossRef]
- Coan, P.M.; Burton, G.J.; Ferguson-Smith, A.C. Imprinted genes in the placenta–A review. Placenta 2005, 26 (Suppl. A), S10–S20. [Google Scholar] [CrossRef]
- Angiolini, E.; Fowden, A.; Coan, P.; Sandovici, I.; Smith, P.; Dean, W.; Burton, G.; Tycko, B.; Reik, W.; Sibley, C.; et al. Regulation of placental efficiency for nutrient transport by imprinted genes. Placenta 2006, 27 (Suppl. A), S98–S102. [Google Scholar] [CrossRef]
- John, R.M. Imprinted genes and the regulation of placental endocrine function: Pregnancy and beyond. Placenta 2017, 56, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Dunwoodie, S.L.; Beddington, R.S. The expression of the imprinted gene Ipl is restricted to extra-embryonic tissues and embryonic lateral mesoderm during early mouse development. Int. J. Dev. Biol. 2002, 46, 459–466. [Google Scholar] [PubMed]
- Qian, N.; Frank, D.; O’Keefe, D.; Dao, D.; Zhao, L.; Yuan, L.; Wang, Q.; Keating, M.; Walsh, C.; Tycko, B. The IPL gene on chromosome 11p15.5 is imprinted in humans and mice and is similar to TDAG51, implicated in Fas expression and apoptosis. Hum. Mol. Genet. 1997, 6, 2021–2029. [Google Scholar] [CrossRef]
- Frank, D.; Fortino, W.; Clark, L.; Musalo, R.; Wang, W.; Saxena, A.; Li, C.M.; Reik, W.; Ludwig, T.; Tycko, B. Placental overgrowth in mice lacking the imprinted gene Ipl. Proc. Natl. Acad. Sci. USA 2002, 99, 7490–7495. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Frank, D.; Panichkul, P.; Van den Veyver, I.B.; Tycko, B.; Thaker, H. The product of the imprinted gene IPL marks human villous cytotrophoblast and is lost in complete hydatidiform mole. Placenta 2003, 24, 835–842. [Google Scholar] [CrossRef]
- Saxena, A.; Morozov, P.; Frank, D.; Musalo, R.; Lemmon, M.A.; Skolnik, E.Y.; Tycko, B. Phosphoinositide binding by the pleckstrin homology domains of Ipl and Tih1. J. Biol. Chem. 2002, 277, 49935–49944. [Google Scholar] [CrossRef] [PubMed]
- Park, C.G.; Lee, S.Y.; Kandala, G.; Lee, S.Y.; Choi, Y. A novel gene product that couples TCR signaling to Fas(CD95) expression in activation-induced cell death. Immunity 1996, 4, 583–591. [Google Scholar] [CrossRef]
- Tunster, S.J.; Creeth, H.D.J.; John, R.M. The imprinted Phlda2 gene modulates a major endocrine compartment of the placenta to regulate placental demands for maternal resources. Dev. Biol. 2016, 409, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.; John, R.; Saxena, A.; Barton, S.; Frank, D.; Fitzpatrick, G.; Higgins, M.J.; Tycko, B. Placental growth retardation due to loss of imprinting of Phlda2. Mech. Dev. 2004, 121, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Tunster, S.J.; Tycko, B.; John, R.M. The imprinted Phlda2 gene regulates extraembryonic energy stores. Mol. Cell. Biol. 2010, 30, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Tunster, S.J.; Van De Pette, M.; John, R.M. Isolating the role of elevated Phlda2 in asymmetric late fetal growth restriction in mice. Dis. Model. Mech. 2014, 7, 1185–1191. [Google Scholar] [CrossRef]
- Apostolidou, S.; Abu-Amero, S.; O’Donoghue, K.; Frost, J.; Olafsdottir, O.; Chavele, K.M.; Whittaker, J.C.; Loughna, P.; Stanier, P.; Moore, G.E. Elevated placental expression of the imprinted PHLDA2 gene is associated with low birth weight. J. Mol. Med. 2007, 85, 379–387. [Google Scholar] [CrossRef]
- McMinn, J.; Wei, M.; Schupf, N.; Cusmai, J.; Johnson, E.B.; Smith, A.C.; Weksberg, R.; Thaker, H.M.; Tycko, B. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta 2006, 27, 540–549. [Google Scholar] [CrossRef]
- Shi, X.; He, Z.; Gao, Y.; Luo, Y.; Gou, C.; Fang, Q. Placental expression of PHLDA2 in selective intrauterine growth restriction in monozygotic twins. Placenta 2014, 35, 428–430. [Google Scholar] [CrossRef]
- Janssen, A.B.; Tunster, S.J.; Heazell, A.E.; John, R.M. Placental PHLDA2 expression is increased in cases of fetal growth restriction following reduced fetal movements. BMC Med. Genet. 2016, 17, 17. [Google Scholar] [CrossRef]
- Ishida, M.; Monk, D.; Duncan, A.J.; Abu-Amero, S.; Chong, J.; Ring, S.M.; Pembrey, M.E.; Hindmarsh, P.C.; Whittaker, J.C.; Stanier, P.; et al. Maternal inheritance of a promoter variant in the imprinted PHLDA2 gene significantly increases birth weight. Am. J. Hum. Genet. 2012, 90, 715–719. [Google Scholar] [CrossRef]
- Coan, P.M.; Ferguson-Smith, A.C.; Burton, G.J. Developmental dynamics of the definitive mouse placenta assessed by stereology. Biol. Reprod. 2004, 70, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.P.; Coan, P.M.; Ferguson-Smith, A.C.; Dean, W.; Hughes, J.; Smith, P.; Reik, W.; Burton, G.J.; Fowden, A.L.; Constância, M. Placental-specific insulin-like growth factor 2 (Igf2) regulates the diffusional exchange characteristics of the mouse placenta. Proc. Natl. Acad. Sci. USA 2004, 101, 8204–8208. [Google Scholar] [CrossRef] [PubMed]
- Constância, M.; Angiolini, E.; Sandovici, I.; Smith, P.; Smith, R.; Kelsey, G.; Dean, W.; Ferguson-Smith, A.; Sibley, C.P.; Reik, W.; et al. Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc. Natl. Acad. Sci. USA 2005, 102, 19219–19224. [Google Scholar] [CrossRef]
- López-Tello, J.; Pérez-García, V.; Khaira, J.; Kusinski, L.C.; Cooper, W.N.; Andreani, A.; Grant, I.; Fernández de Liger, E.; Lam, B.Y.; Hemberger, M.; et al. Fetal and trophoblast PI3K p110α have distinct roles in regulating resource supply to the growing fetus in mice. Elife 2019, 8, e45282. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.D.; Copas, D.K.; Taylor, A.; Landon, M.J.; Stacey, T.E. Permeability of the human placenta in vivo to four non-metabolized hydrophilic molecules. J. Physiol. 1990, 431, 505–513. [Google Scholar] [CrossRef]
- Sibley, C.P. Review article: Mechanisms of ion transfer by the rat placenta: A model for the human placenta? Placenta 1994, 15, 675–691. [Google Scholar] [CrossRef]
- Sibley, C.P.; Brownbill, P.; Glazier, J.D.; Greenwood, S.L. Knowledge needed about the exchange physiology of the placenta. Placenta 2018, 64 (Suppl. 1), S9–S15. [Google Scholar] [CrossRef]
- Kertschanska, S.; Kosanke, G.; Kaufmann, P. Pressure dependence of so-called transtrophoblastic channels during fetal perfusion of human placental villi. Microsc. Res. Tech. 1997, 38, 52–62. [Google Scholar] [CrossRef]
- Brownbill, P.; Mahendran, D.; Owen, D.; Swanson, P.; Thornburg, K.L.; Nelson, D.M.; Sibley, C.P. Denudations as paracellular routes for alphafetoprotein and creatinine across the human syncytiotrophoblast. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R677–R683. [Google Scholar] [CrossRef]
- Stulc, J. Extracellular transport pathways in the haemochorial placenta. Placenta 1989, 10, 113–119. [Google Scholar] [CrossRef]
- Gluckman, P.D. Endocrine and nutritional regulation of prenatal growth. Acta Paediatr. Suppl. 1997, 423, 153–157. [Google Scholar] [CrossRef]
- Carter, A.M. Placental oxygen consumption. Part I: In vivo studies–A review. Placenta 2000, 21 (Suppl. A), S31–S37. [Google Scholar] [CrossRef]
- Vaughan, O.R.; Fowden, A.L. Placental metabolism: Substrate requirements and the response to stress. Reprod. Domest. Anim. 2016, 51 (Suppl. 2), 25–35. [Google Scholar] [CrossRef]
- Hay, W.W., Jr. Regulation of placental metabolism by glucose supply. Reprod. Fertil. Dev. 1995, 7, 365–375. [Google Scholar] [CrossRef]
- Coan, P.M.; Angiolini, E.; Sandovici, I.; Burton, G.J.; Constância, M.; Fowden, A.L. Adaptations in placental nutrient transfer capacity to meet fetal growth demands depend on placental size in mice. J. Physiol. 2008, 586, 4567–4576. [Google Scholar] [CrossRef]
- Coan, P.M.; Vaughan, O.R.; McCarthy, J.; Mactier, C.; Burton, G.J.; Constância, M.; Fowden, A.L. Dietary composition programmes placental phenotype in mice. J. Physiol. 2011, 589, 3659–3670. [Google Scholar] [CrossRef]
- Vaughan, O.R.; Sferruzzi-Perri, A.N.; Fowden, A.L. Maternal corticosterone regulates nutrient allocation to fetal growth in mice. J. Physiol. 2012, 590, 5529–5540. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, A.M.; Camm, E.J.; Sferruzzi-Perri, A.N.; Ashmore, T.J.; Yung, H.W.; Cindrova-Davies, T.; Spiroski, A.M.; Sutherland, M.R.; Logan, A.; Austin-Williams, S.; et al. Placental Adaptation to Early-Onset Hypoxic Pregnancy and Mitochondria-Targeted Antioxidant Therapy in a Rodent Model. Am. J. Pathol. 2018, 188, 2704–2716. [Google Scholar] [CrossRef]
- Sferruzzi-Perri, A.N.; Higgins, J.S.; Vaughan, O.R.; Murray, A.J.; Fowden, A.L. Placental mitochondria adapt developmentally and in response to hypoxia to support fetal growth. Proc. Natl. Acad. Sci. USA 2019, 116, 1621–1626. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A. Maternal constraint of fetal growth and its consequences. Semin. Fetal Neonatal Med. 2004, 9, 419–425. [Google Scholar] [CrossRef]
- Angiolini, E.; Coan, P.M.; Sandovici, I.; Iwajomo, O.H.; Peck, G.; Burton, G.J.; Sibley, C.P.; Reik, W.; Fowden, A.L.; Constância, M. Developmental adaptations to increased fetal nutrient demand in mouse genetic models of Igf2-mediated overgrowth. FASEB J. 2011, 25, 1737–1745. [Google Scholar] [CrossRef]
- Wenzel, P.L.; Leone, G. Expression of Cre recombinase in early diploid trophoblast cells of the mouse placenta. Genesis 2007, 45, 129–134. [Google Scholar] [CrossRef]
- Constância, M.; Hemberger, M.; Hughes, J.; Dean, W.; Ferguson-Smith, A.; Fundele, R.; Stewart, F.; Kelsey, G.; Fowden, A.; Sibley, C.; et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002, 417, 945–948. [Google Scholar] [CrossRef]
- Coan, P.M.; Fowden, A.L.; Constância, M.; Ferguson-Smith, A.C.; Burton, G.J.; Sibley, C.P. Disproportional effects of Igf2 knockout on placental morphology and diffusional exchange characteristics in the mouse. J. Physiol. 2008, 586, 5023–5032. [Google Scholar] [CrossRef] [PubMed]
- Sekita, Y.; Wagatsuma, H.; Nakamura, K.; Ono, R.; Kagami, M.; Wakisaka, N.; Hino, T.; Suzuki-Migishima, R.; Kohda, T.; Ogura, A.; et al. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat. Genet. 2008, 40, 243–248. [Google Scholar] [CrossRef]
- Patten, M.M.; Cowley, M.; Oakey, R.J.; Feil, R. Regulatory links between imprinted genes: Evolutionary predictions and consequences. Proc. Biol. Sci. 2016, 283, 20152760. [Google Scholar] [CrossRef]


{kind=link}
| Gestational Age | Genotype | N | Fetal Wet Weight (g) ± SE | Placental Wet Weight (g) ± SE | F/P Weight Ratios ± SE |
|---|---|---|---|---|---|
| E16 | Phlda2+/+ | 48 | 0.398 ± 0.005 | 0.100 ± 0.002 | 4.007 ± 0.083 |
| Phlda2−/+ | 49 | 0.384 ± 0.005 | 0.129 ± 0.002 | 3.099 ± 0.081 | |
| (% of Phlda2+/+) | 96.4 * | 129.7 *** | 77.3 *** | ||
| E19 | Phlda2+/+ | 88 | 1.079 ± 0.009 | 0.082 ± 0.001 | 13.395 ± 0.227 |
| Phlda2−/+ | 94 | 1.069 ± 0.009 | 0.106 ± 0.001 | 10.247 ± 0.172 | |
| (% of Phlda2+/+) | 99.1 | 129.3 *** | 76.5 *** |
| E16 | E19 | |||||
|---|---|---|---|---|---|---|
| Phlda2+/+ | Phlda2−/+ | % of Phlda2+/+ | Phlda2+/+ | Phlda2−/+ | % of Phlda2+/+ | |
| Placenta | 93.49 ± 3.215 | 139.36 ± 5.754 | 149 ** | 88.73 ± 7.377 | 116.41 ± 1.677 | 131 * |
| Lz | 40.71 ± 2.151 | 44.89 ± 1.575 | 110 | 44.13 ± 5.668 | 50.60 ± 2.122 | 115 |
| Jz | 32.14 ± 0.172 | 71.31 ± 8.042 | 222 ** | 30.54 ± 1.176 | 47.26 ± 2.597 | 155 ** |
| Db | 18.13 ± 1.499 | 19.78 ± 4.538 | 109 | 11.07 ± 1.603 | 13.82 ± 1.535 | 125 |
| Jz/Lz | 0.80 ± 0.049 | 1.58 ± 0.138 | 198 ** | 0.73 ± 0.110 | 0.94 ± 0.081 | 129 |
| E16 | E19 | |||||
|---|---|---|---|---|---|---|
| Phlda2+/+ | Phlda2−/+ | % of Phlda2+/+ | Phlda2+/+ | Phlda2−/+ | % of Phlda2+/+ | |
| LT | 26.07 ± 0.139 | 29.57 ± 1.418 | 113 | 28.92 ± 5.013 | 30.80 ± 1.059 | 107 |
| MBS | 7.37 ± 1.291 | 7.86 ± 1.156 | 107 | 7.47 ± 0.754 | 11.27 ± 0.938 | 151 * |
| FC | 7.27 ± 1.384 | 7.45 ± 0.154 | 103 | 7.73 ± 1.565 | 8.52 ±1.469 | 110 |
| MBS SA | 18.40 ± 0.929 | 16.71 ± 2.075 | 91 | 22.28 ± 2.848 | 27.57 ± 2.182 | 124 |
| FC SA | 14.97 ± 1.351 | 15.78 ± 1.281 | 105 | 16.65 ± 3.058 | 19.86 ± 3.718 | 119 |
| IMT | 4.29 ± 0.373 | 4.25 ± 0.154 | 99 | 3.99 ± 0.185 | 4.08 ± 0.295 | 102 |
| TDC | 0.0069 ± 0.0006 | 0.0068 ± 0.0008 | 99 | 0.0085 ± 0.0011 | 0.0104 ± 0.0019 | 123 |
| SDC | 0.0205 ± 0.0020 | 0.0200 ± 0.0033 | 98 | 0.0072 ± 0.0007 | 0.0092 ± 0.0020 | 129 |
| Gene Deletion | Theoretical Diffusion Capacity | Passive Permeability | Transporter Mediated Transport | Placental Size | Normal Role of Gene on Passive Permeability | Reference | ||
|---|---|---|---|---|---|---|---|---|
| MeAIB | Glucose | Lz | Jz | |||||
| Phlda2 | No Δ | ↑ (inulin) | No Δ | No Δ | No Δ | ↑ | Restricts paracellular pore length, radius or number | This study |
| Igf2P0 | ↓ | ↓ (inulin, EDTA, mannitol) | No Δ | ↑ | ↓ | ↓ | Increases TDC by reducing interhemal membrane thickness and increasing surface area | [23,24] |
| Igf2 | ↓ | ↓ (inulin) | ↓ | No Δ | ↓ | ↓ | Increases TDC by reducing interhemal membrane thickness and increasing surface area | [24,45] |
| H19Δ13 (leading to Igf2 LOI) | ↑ | ↓ (inulin, mannitol) | ↓ | ↓ | ↑ | ↑ | Increases paracellular pore characteristics and reduces TDC through effects on surface area | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angiolini, E.; Sandovici, I.; Coan, P.M.; Burton, G.J.; Sibley, C.P.; Fowden, A.L.; Constância, M. Deletion of the Imprinted Phlda2 Gene Increases Placental Passive Permeability in the Mouse. Genes 2021, 12, 639. https://doi.org/10.3390/genes12050639
Angiolini E, Sandovici I, Coan PM, Burton GJ, Sibley CP, Fowden AL, Constância M. Deletion of the Imprinted Phlda2 Gene Increases Placental Passive Permeability in the Mouse. Genes. 2021; 12(5):639. https://doi.org/10.3390/genes12050639
Chicago/Turabian StyleAngiolini, Emily, Ionel Sandovici, Philip M. Coan, Graham J. Burton, Colin P. Sibley, Abigail L. Fowden, and Miguel Constância. 2021. "Deletion of the Imprinted Phlda2 Gene Increases Placental Passive Permeability in the Mouse" Genes 12, no. 5: 639. https://doi.org/10.3390/genes12050639
APA StyleAngiolini, E., Sandovici, I., Coan, P. M., Burton, G. J., Sibley, C. P., Fowden, A. L., & Constância, M. (2021). Deletion of the Imprinted Phlda2 Gene Increases Placental Passive Permeability in the Mouse. Genes, 12(5), 639. https://doi.org/10.3390/genes12050639