
The PARP Way to Epigenetic Changes


Abstract
1. Introduction
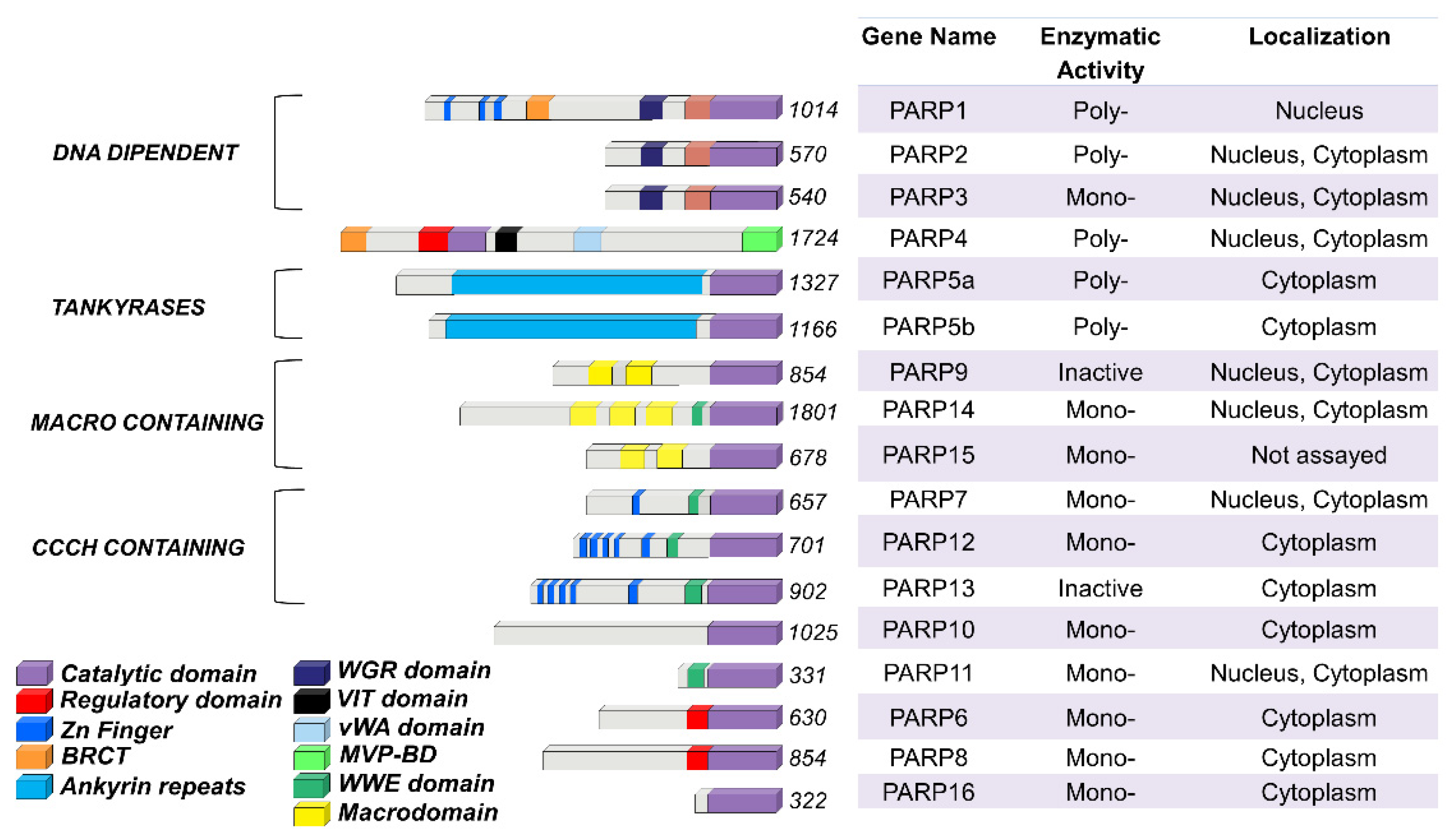
2. The PARP Family Members
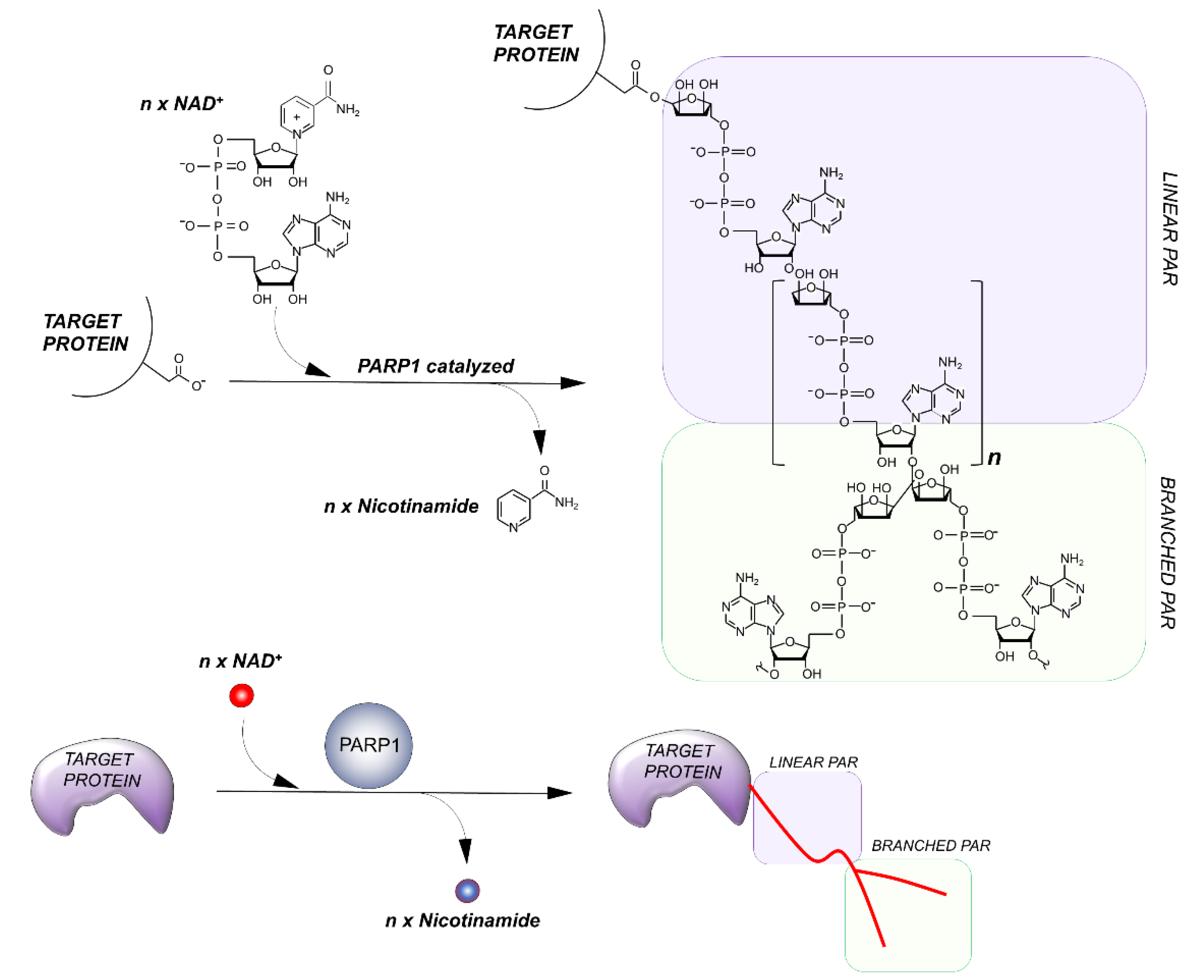
3. Basic Notions of PARylation
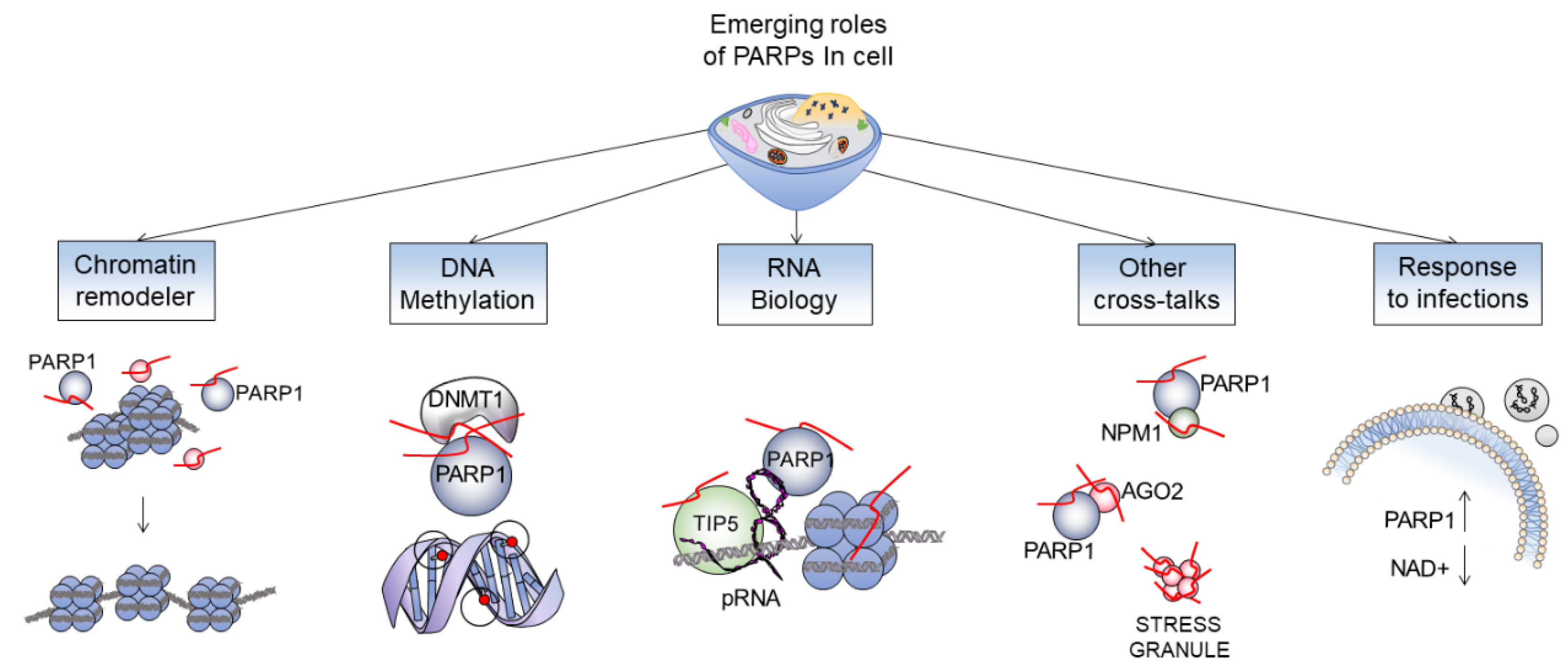
4. Getting Closer to the Edge of Unexplained PARP Functions
5. Remodeling of Chromatin: Can Histones Be PARylated?
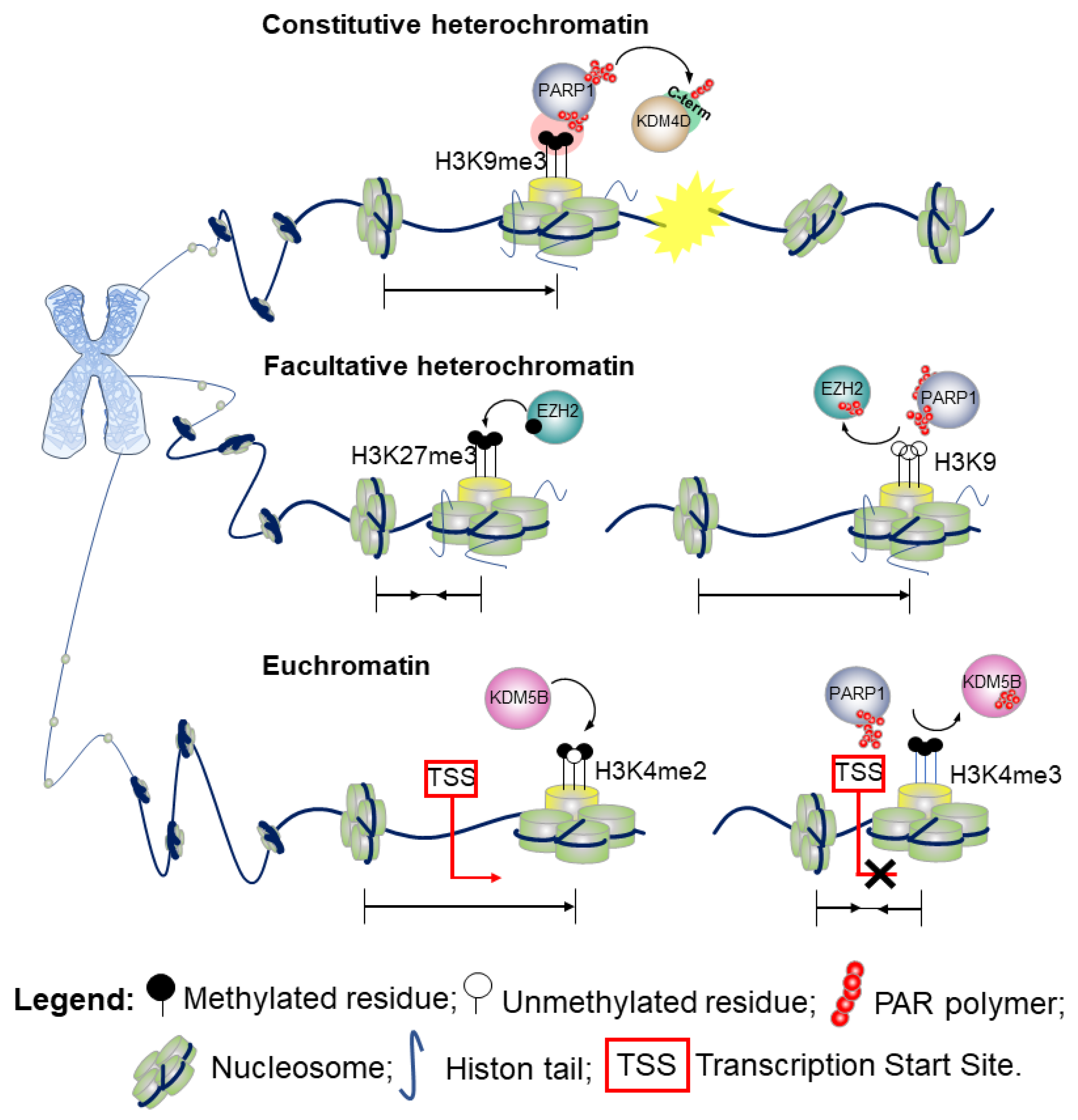
6. PARP1 as an Effector of Chromatin Modifications
- a.
- Constitutive heterochromatin, highly condensed regions of DNA that display species-specific genomic coverage and variability ranging from 30% to 90%;
- b.
- Facultative heterochromatin, regions of packaged DNA that can be reverted to euchromatin upon specific conditions and histone modifications, accounting for 45% of the genome;
- c.
- Euchromatin, highly accessible and decondensed portions of the DNA that are transcriptionally active.
- a.
- Constitutive heterochromatinH3 lysine 9 trimethylation (H3K9me3) is the hallmark of highly condensed chromatin. Defects in PARylation are commonly associated with loss of the methylation marker at the centromeric heterochromatin of pericentromeric regions. While the di-(me2) and tri-methylation forms of H3K9 are enriched at the transcriptional start site (TSS) of silenced genes, the mono-methylation variant (H3K9me1) marks promoters of actively transcribed genes [35]. Although H3K9me3 aids the recruitment of chromatin enzymes involved in the DDR, its presence impairs the DNA repairing process that requires a decondensed state of chromatin to enable the action of the DNA-repair effectors [36]. PARylation of the lysine-specific demethylase 4D (KDM4D) [37] at the C-terminal domain engages KDM4D to the sites of DNA damage. By promoting the demethylation of H3K9, PARylation reduces the degree of chromatin compaction, thus playing a key role in the propagation of DDR in vivo [37,38]. A schematic outlining this molecular mechanism is shown in Figure 4 (upper part). Further evidence show that in response to DNA damage when the chromatin undergoes structural reorganization to ensure accurate DNA repair [3], PARP1 not only promotes recruitment of proteins at the damaged site, but also acts as a chromatin remodeler to facilitate the access of the DNA repair machinery. Indeed, several studies have demonstrated that PARylation of the histones causes chromatin decondensation [25,39].For instance, by recruiting chromodomain helicase DNA binding protein 2 (CHD2) at DSBs, PARP1 triggers deposition of the histone variant H3.3, and ultimately chromatin relaxation thereby regulating the assembly of non-homologous end-joining (NHEJ) complexes to rescue genomic integrity.Therefore, PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by NEHJ. In the NHEJ pathway, PARP1 may also serve as a scaffold to recruit at sites of DNA damage a number of transcription repression complexes, including the nucleosome remodeler and deacetylase (NuRD), the complex proteins CHD4, the metastasis-associated protein 1 (MTA1) [40,41], and members of Polycomb repressive complex 1 (PRC1) [40].
- b.
- Facultative heterochromatinA positional effect of PARylation has also been proposed in the context of facultative heterochromatin as observed for the H3K9me3/2 demethylase KDM4D [42]. Indeed, PARylation of KDM4D conserved N-terminal domain—the JmjN, that is a substrates for PARP-1, inhibits its activity at the promoter of retinoic acid receptor (RAR)-dependent genes thereby resulting in transcriptional repression [42]. Hence, whilst PARylation at the C-terminal domain promotes KDM4D demethylase activity and reduces the degree of chromatin compaction [37], PARylation at the N-terminal end results in an opposite effect. Alternatively, a model wherein PARP1 cooperates in the establishment of the heterochromatin landscape upon inhibition of KDM4D has also been proposed [42]. This action of PARP1 can be reversed by poly-ADP-ribose glycohydrolase (PARG), the catabolic enzyme that cleaves the ADP-ribose polymers synthesized by PARP1. PARG counteracts the action of PARP1 and favors an open structure of the chromatin, promoting an active transcriptional state [42].In the attempt to further understand the PARP1 and PARylation conundrum, a 2015 study investigated the effects of PARP1 on global gene expression in a lymphoblastoid B cell line [43]. These data revealed that PARylation controls the methyltransferase enhancer of zeste homolog 2 (EZH2), the catalytic subunit of the polycomb repressive complex 2 (PRC2). PRC2 is responsible for the trimethylation of the lysine 27 on histone 3 (H3K27me3), which leads to chromatin compaction and gene silencing. A schematic example of the molecular mechanism is presented in Figure 4 (middle part).Upon pharmacological inhibition of PARP and shRNA-mediated downregulation of PARP1, EZH2 expression is induced, resulting in increased global H3K27me3 [43].Furthermore, PARP activity is required for retaining PRC2, the supporting protein suppressor of Zeste 12 (SUZ12) and the embryonic ectoderm development (EED), at the site of DNA damage. Surprisingly, EZH2 is not recruited directly by single-strand breaks or UV damage [44].
- c.
- EuchromatinTwo methylation states of H3 lysine 4 (H3K4me2/me3) are enriched at the TSS of actively transcribed genes and correspond to euchromatic regions in the genome. The monomethylation state typically marks enhancers [45]. As shown in Figure 4 (bottom part), PARylation impairs the enzymatic activity of KDM5B, a histone lysine demethylase of the H3 trimethylated lysine 4, and the respective binding to H3 in in vitro assays. Consistently, inhibition of PARylation in vivo results in increased levels of KDM5B at the TSS of active genes and decreased levels of H3K4me3.The interplay between PARP1 and KDM5B has been considered a regulatory mechanism to control the chromatin state at the basal and signal-transcriptional level [27]. While PARylation recruits KDM5B to DNA damaged sites, demethylation of H3K4me3 in proximity to DNA breaks helps to recruit proteins involved in the DNA-damage repair, including BRCA1 [46]. Hence, PARylation of KDM5B could have a double effect on chromatin association. Remodeling of the chromatin during DSB repair can include variation on the usual mechanism observed in gene transcription, including the physical movement of nucleosomes, histone variant exchange, and dynamic changes in histone acetylation and methylation to create nucleosome-free regions that facilitate the entire repair process [47]. A recent finding from Gong et al. using live imaging, revealed that PARP1 recruits KDM5A through PAR chains at the damaged chromatin side, leading to rapid erasure of H3K4me3 and promoting recruitment of a second repair protein, ZMYND8 [48]. Consistent with these findings, loss of KDM5A attenuates the normal drop in local transcriptional activity adjacent to DSBs, in line with loading of ZMYND8 (and loss of H3K4me3) acting as a general transcriptional repressor [48].Further, PARP1–3 proteins can directly PARylate DNA breaks by loading PAR units to terminal phosphates. [34].Finally, a 2019 study details a fascinating interplay between PARP1, chromatin, and RNA polymerase II (RNAPII). It was found that RNAPII pauses elongation when it encounters PARP1 bound to chromatin. Knockout of the PARP1 gene prevented this pause from occurring, implicating that PARP1 plays a regulatory role in chromatin changes and transcription [49].
7. PARP1 Modulates the Delicate Balance of DNA Methylation
8. A PARP1 RNA Interplay
9. Mechanisms and Clinical Applications of PARP Inhibitors
10. Other Cross-Talks in the Complex Network Created by PARP1
11. Beyond Cancer Treatment: A Novel Target for COVID-19?
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: Progress and puzzles. Mol. Asp. Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef]
- Caiafa, P.; Guastafierro, T.; Zampieri, M. Epigenetics: Poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J. 2009, 23, 672–678. [Google Scholar] [CrossRef]
- Palazzo, L.; Mikolcevic, P.; Mikoc, A.; Ahel, I. ADP-ribosylation signalling and human disease. Open Biol. 2019, 9, 190041. [Google Scholar] [CrossRef]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Menissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Chi, N.W. Identification of a tankyrase-binding motif shared by IRAP, TAB182, and human TRF1 but not mouse TRF1. NuMA contains this RXXPDG motif and is a novel tankyrase partner. J. Biol. Chem. 2002, 277, 31887–31892. [Google Scholar] [CrossRef]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- Sugimura, T.; Miwa, M. Poly(ADP-ribose): Historical perspective. Mol. Cell. Biochem. 1994, 138, 5–12. [Google Scholar] [CrossRef]
- Zandarashvili, L.; Langelier, M.F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Molinete, M.; Boeuf, H.; de Murcia, G.; Menissier-de Murcia, J. The human poly(ADP-ribose) polymerase nuclear localization signal is a bipartite element functionally separate from DNA binding and catalytic activity. EMBO J. 1992, 11, 3263–3269. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Ruf, A.; Mennissier de Murcia, J.; de Murcia, G.; Schulz, G.E. Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc. Natl. Acad. Sci. USA 1996, 93, 7481–7485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Ding, M.; Yu, Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods 2013, 10, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Kiehlbauch, C.C.; Aboul-Ela, N.; Jacobson, E.L.; Ringer, D.P.; Jacobson, M.K. High resolution fractionation and characterization of ADP-ribose polymers. Anal. Biochem. 1993, 208, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Loseva, O.; Jemth, A.S.; Bryant, H.E.; Schuler, H.; Lehtio, L.; Karlberg, T.; Helleday, T. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J. Biol. Chem. 2010, 285, 8054–8060. [Google Scholar] [CrossRef]
- Kleine, H.; Poreba, E.; Lesniewicz, K.; Hassa, P.O.; Hottiger, M.O.; Litchfield, D.W.; Shilton, B.H.; Luscher, B. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol. Cell 2008, 32, 57–69. [Google Scholar] [CrossRef]
- Dulaney, C.; Marcrom, S.; Stanley, J.; Yang, E.S. Poly(ADP-ribose) polymerase activity and inhibition in cancer. Semin. Cell. Dev. Biol. 2017, 63, 144–153. [Google Scholar] [CrossRef]
- Reale, A.; Matteis, G.D.; Galleazzi, G.; Zampieri, M.; Caiafa, P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene 2005, 24, 13–19. [Google Scholar] [CrossRef]
- Guetg, C.; Scheifele, F.; Rosenthal, F.; Hottiger, M.O.; Santoro, R. Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA. Mol. Cell 2012, 45, 790–800. [Google Scholar] [CrossRef]
- Badawy, A.A. Immunotherapy of COVID-19 with poly (ADP-ribose) polymerase inhibitors: Starting with nicotinamide. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Aubin, R.J.; Frechette, A.; de Murcia, G.; Mandel, P.; Lord, A.; Grondin, G.; Poirier, G.G. Correlation between endogenous nucleosomal hyper(ADP-ribosyl)ation of histone H1 and the induction of chromatin relaxation. EMBO J. 1983, 2, 1685–1693. [Google Scholar] [CrossRef]
- Quenet, D.; El Ramy, R.; Schreiber, V.; Dantzer, F. The role of poly(ADP-ribosyl)ation in epigenetic events. Int. J. Biochem. Cell. Biol. 2009, 41, 60–65. [Google Scholar] [CrossRef]
- Messner, S.; Altmeyer, M.; Zhao, H.; Pozivil, A.; Roschitzki, B.; Gehrig, P.; Rutishauser, D.; Huang, D.; Caflisch, A.; Hottiger, M.O. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010, 38, 6350–6362. [Google Scholar] [CrossRef]
- Kim, M.Y.; Mauro, S.; Gevry, N.; Lis, J.T.; Kraus, W.L. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 2004, 119, 803–814. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell 2010, 39, 736–749. [Google Scholar] [CrossRef]
- Wright, R.H.; Castellano, G.; Bonet, J.; Le Dily, F.; Font-Mateu, J.; Ballare, C.; Nacht, A.S.; Soronellas, D.; Oliva, B.; Beato, M. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Dev. 2012, 26, 1972–1983. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Chen, Y.; Wu, J.; Chen, S.H.; Liu, X.; Singh, A.K.; Yu, X. Poly(ADP-ribosyl)ation mediates early phase histone eviction at DNA lesions. Nucleic Acids Res. 2020, 48, 3001–3013. [Google Scholar] [CrossRef]
- Pinnola, A.; Naumova, N.; Shah, M.; Tulin, A.V. Nucleosomal core histones mediate dynamic regulation of poly(ADP-ribose) polymerase 1 protein binding to chromatin and induction of its enzymatic activity. J. Biol. Chem. 2007, 282, 32511–32519. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell. Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Muthurajan, U.M.; Hepler, M.R.; Hieb, A.R.; Clark, N.J.; Kramer, M.; Yao, T.; Luger, K. Automodification switches PARP-1 function from chromatin architectural protein to histone chaperone. Proc. Natl. Acad. Sci. USA 2014, 111, 12752–12757. [Google Scholar] [CrossRef] [PubMed]
- Gibbs-Seymour, I.; Fontana, P.; Rack, J.G.M.; Ahel, I. HPF1/C4orf27 Is a PARP-1-Interacting Protein that Regulates PARP-1 ADP-Ribosylation Activity. Mol. Cell 2016, 62, 432–442. [Google Scholar] [CrossRef]
- Matta, E.; Kiribayeva, A.; Khassenov, B.; Matkarimov, B.T.; Ishchenko, A.A. Insight into DNA substrate specificity of PARP1-catalysed DNA poly(ADP-ribosyl)ation. Sci. Rep. 2020, 10, 3699. [Google Scholar] [CrossRef]
- Krishnan, S.; Horowitz, S.; Trievel, R.C. Structure and function of histone H3 lysine 9 methyltransferases and demethylases. Chembiochem 2011, 12, 254–263. [Google Scholar] [CrossRef]
- Cann, K.L.; Dellaire, G. Heterochromatin and the DNA damage response: The need to relax. Biochem. Cell Biol. 2011, 89, 45–60. [Google Scholar] [CrossRef]
- Khoury-Haddad, H.; Guttmann-Raviv, N.; Ipenberg, I.; Huggins, D.; Jeyasekharan, A.D.; Ayoub, N. PARP1-dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E728–E737. [Google Scholar] [CrossRef] [PubMed]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef] [PubMed]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiacovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010, 29, 3130–3139. [Google Scholar] [CrossRef]
- Le May, N.; Iltis, I.; Ame, J.C.; Zhovmer, A.; Biard, D.; Egly, J.M.; Schreiber, V.; Coin, F. Poly (ADP-ribose) glycohydrolase regulates retinoic acid receptor-mediated gene expression. Mol. Cell 2012, 48, 785–798. [Google Scholar] [CrossRef]
- Martin, K.A.; Cesaroni, M.; Denny, M.F.; Lupey, L.N.; Tempera, I. Global Transcriptome Analysis Reveals That Poly(ADP-Ribose) Polymerase 1 Regulates Gene Expression through EZH2. Mol. Cell. Biol. 2015, 35, 3934–3944. [Google Scholar] [CrossRef]
- Campbell, S.; Ismail, I.H.; Young, L.C.; Poirier, G.G.; Hendzel, M.J. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle 2013, 12, 2675–2683. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Yang, S.; Song, N.; Zhou, X.; Gao, J.; Yu, N.; Shan, L.; Wang, Q.; Liang, J.; et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. USA 2014, 111, 7096–7101. [Google Scholar] [CrossRef]
- Gursoy-Yuzugullu, O.; House, N.; Price, B.D. Patching Broken DNA: Nucleosome Dynamics and the Repair of DNA Breaks. J. Mol. Biol. 2016, 428, 1846–1860. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Clouaire, T.; Aguirrebengoa, M.; Legube, G.; Miller, K.M. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell Biol. 2017, 216, 1959–1974. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Al-Tinawi, Q.M.H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Coupling of PARP1-mediated chromatin structural changes to transcriptional RNA polymerase II elongation and cotranscriptional splicing. Epigenetics Chromatin 2019, 12, 15. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.T. DNA methylation remodeling in vitro and in vivo. Curr. Opin. Genet. Dev. 2015, 34, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Dunbar, A.; Gondek, L.P.; Mohan, S.; Rataul, M.; O’Keefe, C.; Sekeres, M.; Saunthararajah, Y.; Maciejewski, J.P. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 2009, 113, 1315–1325. [Google Scholar] [CrossRef]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression--belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- de Capoa, A.; Febbo, F.R.; Giovannelli, F.; Niveleau, A.; Zardo, G.; Marenzi, S.; Caiafa, P. Reduced levels of poly(ADP-ribosyl)ation result in chromatin compaction and hypermethylation as shown by cell-by-cell computer-assisted quantitative analysis. FASEB J. 1999, 13, 89–93. [Google Scholar] [CrossRef]
- Zardo, G.; D’Erme, M.; Reale, A.; Strom, R.; Perilli, M.; Caiafa, P. Does poly(ADP-ribosyl)ation regulate the DNA methylation pattern? Biochemistry 1997, 36, 7937–7943. [Google Scholar] [CrossRef]
- Zampieri, M.; Guastafierro, T.; Calabrese, R.; Ciccarone, F.; Bacalini, M.G.; Reale, A.; Perilli, M.; Passananti, C.; Caiafa, P. ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem. J. 2012, 441, 645–652. [Google Scholar] [CrossRef]
- Caiafa, P.; Zlatanova, J. CCCTC-binding factor meets poly(ADP-ribose) polymerase-1. J. Cell. Physiol. 2009, 219, 265–270. [Google Scholar] [CrossRef]
- Caiafa, P.; Zampieri, M. DNA methylation and chromatin structure: The puzzling CpG islands. J. Cell. Biochem. 2005, 94, 257–265. [Google Scholar] [CrossRef]
- Kemp, C.J.; Moore, J.M.; Moser, R.; Bernard, B.; Teater, M.; Smith, L.E.; Rabaia, N.A.; Gurley, K.E.; Guinney, J.; Busch, S.E.; et al. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 2014, 7, 1020–1029. [Google Scholar] [CrossRef]
- Guastafierro, T.; Cecchinelli, B.; Zampieri, M.; Reale, A.; Riggio, G.; Sthandier, O.; Zupi, G.; Calabrese, L.; Caiafa, P. CCCTC-binding factor activates PARP-1 affecting DNA methylation machinery. J. Biol. Chem. 2008, 283, 21873–21880. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Ginjala, V.; Pant, V.; Chernukhin, I.; Whitehead, J.; Docquier, F.; Farrar, D.; Tavoosidana, G.; Mukhopadhyay, R.; Kanduri, C.; et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat. Genet. 2004, 36, 1105–1110. [Google Scholar] [CrossRef]
- Nalabothula, N.; Al-jumaily, T.; Eteleeb, A.M.; Flight, R.M.; Xiaorong, S.; Moseley, H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Genome-Wide Profiling of PARP1 Reveals an Interplay with Gene Regulatory Regions and DNA Methylation. PLoS ONE 2015, 10, e0135410. [Google Scholar] [CrossRef]
- Li, M.; Cascino, P.; Ummarino, S.; Di Ruscio, A. Application of Induced Pluripotent Stem Cell Technology to the Study of Hematological Diseases. Cells 2017, 6, 7. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Doege, C.A.; Inoue, K.; Yamashita, T.; Rhee, D.B.; Travis, S.; Fujita, R.; Guarnieri, P.; Bhagat, G.; Vanti, W.B.; Shih, A.; et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 2012, 488, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Hajkova, P.; Jeffries, S.J.; Lee, C.; Miller, N.; Jackson, S.P.; Surani, M.A. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science 2010, 329, 78–82. [Google Scholar] [CrossRef]
- Mayer, C.; Schmitz, K.M.; Li, J.; Grummt, I.; Santoro, R. Intergenic transcripts regulate the epigenetic state of rRNA genes. Mol. Cell 2006, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Schmitz, K.M.; Sandoval, J.; Grummt, I. Intergenic transcripts originating from a subclass of ribosomal DNA repeats silence ribosomal RNA genes in trans. EMBO Rep. 2010, 11, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Melikishvili, M.; Chariker, J.H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Transcriptome-wide identification of the RNA-binding landscape of the chromatin-associated protein PARP1 reveals functions in RNA biogenesis. Cell Discov. 2017, 3, 17043. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, D.; Huang, D.; Song, H.; Mei, H.; Fang, E.; Wang, X.; Yang, F.; Zheng, L.; Huang, K.; et al. Risk-Associated Long Noncoding RNA FOXD3-AS1 Inhibits Neuroblastoma Progression by Repressing PARP1-Mediated Activation of CTCF. Mol. Ther. 2018, 26, 755–773. [Google Scholar] [CrossRef]
- Leung, A.K.; Vyas, S.; Rood, J.E.; Bhutkar, A.; Sharp, P.A.; Chang, P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol. Cell 2011, 42, 489–499. [Google Scholar] [CrossRef]
- Bock, F.J.; Todorova, T.T.; Chang, P. RNA Regulation by Poly(ADP-Ribose) Polymerases. Mol. Cell 2015, 58, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Im, S.A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 1700. [Google Scholar] [CrossRef]
- Del Campo, J.M.; Matulonis, U.A.; Malander, S.; Provencher, D.; Mahner, S.; Follana, P.; Waters, J.; Berek, J.S.; Woie, K.; Oza, A.M.; et al. Niraparib Maintenance Therapy in Patients With Recurrent Ovarian Cancer After a Partial Response to the Last Platinum-Based Chemotherapy in the ENGOT-OV16/NOVA Trial. J. Clin. Oncol. 2019, 37, 2968–2973. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Rucaparib: A Review in Ovarian Cancer. Target. Oncol. 2019, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Jadhav, H.; Kothari, A.; Liu, R.; Guerriero, J.L.; Shapiro, G.I. STING agonism enhances anti-tumor immune responses and therapeutic efficacy of PARP inhibition in BRCA-associated breast cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980 e2975. [Google Scholar] [CrossRef] [PubMed]
- Bochum, S.; Berger, S.; Martens, U.M. Olaparib. Recent Results Cancer Res. 2018, 211, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.; Wang, Y.; Mukhopadhyay, D.; Backlund, P.; Kolli, N.; Yergey, A.; Wilkinson, K.D.; Dasso, M. Nucleolar protein B23/nucleophosmin regulates the vertebrate SUMO pathway through SENP3 and SENP5 proteases. J. Cell. Biol. 2008, 183, 589–595. [Google Scholar] [CrossRef]
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279. [Google Scholar] [CrossRef]
- Meder, V.S.; Boeglin, M.; de Murcia, G.; Schreiber, V. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J. Cell. Sci. 2005, 118, 211–222. [Google Scholar] [CrossRef]
- Chan, Y.A.; Aristizabal, M.J.; Lu, P.Y.; Luo, Z.; Hamza, A.; Kobor, M.S.; Stirling, P.C.; Hieter, P. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 2014, 10, e1004288. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Koike, A.; Nishikawa, H.; Wu, W.; Okada, Y.; Venkitaraman, A.R.; Ohta, T. Recruitment of phosphorylated NPM1 to sites of DNA damage through RNF8-dependent ubiquitin conjugates. Cancer Res. 2010, 70, 6746–6756. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef]
- Anglada, T.; Genesca, A.; Martin, M. Age-associated deficient recruitment of 53BP1 in G1 cells directs DNA double-strand break repair to BRCA1/CtIP-mediated DNA-end resection. Aging 2020, 12, 24872–24893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lou, L.; Peng, B.; Song, X.; Reizes, O.; Almasan, A.; Gong, Z. Nudix Hydrolase NUDT16 Regulates 53BP1 Protein by Reversing 53BP1 ADP-Ribosylation. Cancer Res. 2020, 80, 999–1010. [Google Scholar] [CrossRef]
- Valenti, G.; Quinn, H.M.; Heynen, G.; Lan, L.; Holland, J.D.; Vogel, R.; Wulf-Goldenberg, A.; Birchmeier, W. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017, 77, 2134–2147. [Google Scholar] [CrossRef]
- De Vos, M.; El Ramy, R.; Quenet, D.; Wolf, P.; Spada, F.; Magroun, N.; Babbio, F.; Schreiber, V.; Leonhardt, H.; Bonapace, I.M.; et al. Poly(ADP-ribose) polymerase 1 (PARP1) associates with E3 ubiquitin-protein ligase UHRF1 and modulates UHRF1 biological functions. J. Biol. Chem. 2014, 289, 16223–16238. [Google Scholar] [CrossRef] [PubMed]
- Hahm, J.Y.; Kang, J.Y.; Park, J.W.; Jung, H.; Seo, S.B. Methylated-UHRF1 and PARP1 interaction is critical for homologous recombination. BMB Rep. 2020, 53, 112–117. [Google Scholar] [CrossRef]
- Martin-Hernandez, K.; Rodriguez-Vargas, J.M.; Schreiber, V.; Dantzer, F. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Semin. Cell. Dev. Biol. 2017, 63, 92–101. [Google Scholar] [CrossRef]
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell. Dev. Biol. 2017, 63, 123–134. [Google Scholar] [CrossRef]
- Ohn, T.; Anderson, P. The role of posttranslational modifications in the assembly of stress granules. Wiley Interdiscip Rev. RNA 2010, 1, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Challa, S.; Jones, A.; Kraus, W.L. PARPs and ADP-ribosylation in RNA biology: From RNA expression and processing to protein translation and proteostasis. Genes Dev. 2020, 34, 302–320. [Google Scholar] [CrossRef]
- Szabo, C.; Martins, V.; Liaudet, L. Poly(ADP-Ribose) Polymerase Inhibition in Acute Lung Injury. A Reemerging Concept. Am. J. Respir. Cell. Mol. Biol. 2020, 63, 571–590. [Google Scholar] [CrossRef] [PubMed]
- Heer, C.D.; Sanderson, D.J.; Voth, L.S.; Alhammad, Y.M.O.; Schmidt, M.S.; Trammell, S.A.J.; Perlman, S.; Cohen, M.S.; Fehr, A.R.; Brenner, C. Coronavirus infection and PARP expression dysregulate the NAD metabolome: An actionable component of innate immunity. J. Biol. Chem. 2020, 295, 17986–17996. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.; Banyai, K.; Thaventhiran, J.; Le Quesne, J.; Helyes, Z.; Bai, P. Repositioning PARP inhibitors for SARS-CoV-2 infection(COVID-19); a new multi-pronged therapy for acute respiratory distress syndrome? Br. J. Pharm. 2020, 177, 3635–3645. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Reference |
|---|---|---|
| Olaparib | In HER-2 negative metastatic breast cancer patients with a germline BRCA mutation, olaparib has been shown to be very effective. Response rate of 59.9% compared to 28.8% in the standard therapy group. | [77]. |
| Niraparib | In patients with platinum sensitive recurrent ovarian cancer, niraparib greatly enhanced progression-free survival as compared to placebo. These results were consistent regardless of a germline BRCA mutation or homologous recombination deficiency (HRD) status. | [78] |
| Rucaparib | Rucaparib is generally a third (or later) line treatment used in patients with BRCA mutated ovarian cancer and as maintenance therapy for patients with recurrent or relapsed platinum sensitive ovarian cancer. Analysis has revealed an objective response rate of 54%. | [79] |
| Talazoparib | Used in patients with advanced breast cancer and germline BRCA mutations. Talazoparib has shown a significantly higher likelihood of progression-free survival (62.6% compared to 27.2% in the standard therapy group). | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ummarino, S.; Hausman, C.; Di Ruscio, A. The PARP Way to Epigenetic Changes. Genes 2021, 12, 446. https://doi.org/10.3390/genes12030446
Ummarino S, Hausman C, Di Ruscio A. The PARP Way to Epigenetic Changes. Genes. 2021; 12(3):446. https://doi.org/10.3390/genes12030446
Chicago/Turabian StyleUmmarino, Simone, Clinton Hausman, and Annalisa Di Ruscio. 2021. "The PARP Way to Epigenetic Changes" Genes 12, no. 3: 446. https://doi.org/10.3390/genes12030446
APA StyleUmmarino, S., Hausman, C., & Di Ruscio, A. (2021). The PARP Way to Epigenetic Changes. Genes, 12(3), 446. https://doi.org/10.3390/genes12030446