
Identification of Chlorophyll Metabolism- and Photosynthesis-Related Genes Regulating Green Flower Color in Chrysanthemum by Integrative Transcriptome and Weighted Correlation Network Analyses

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
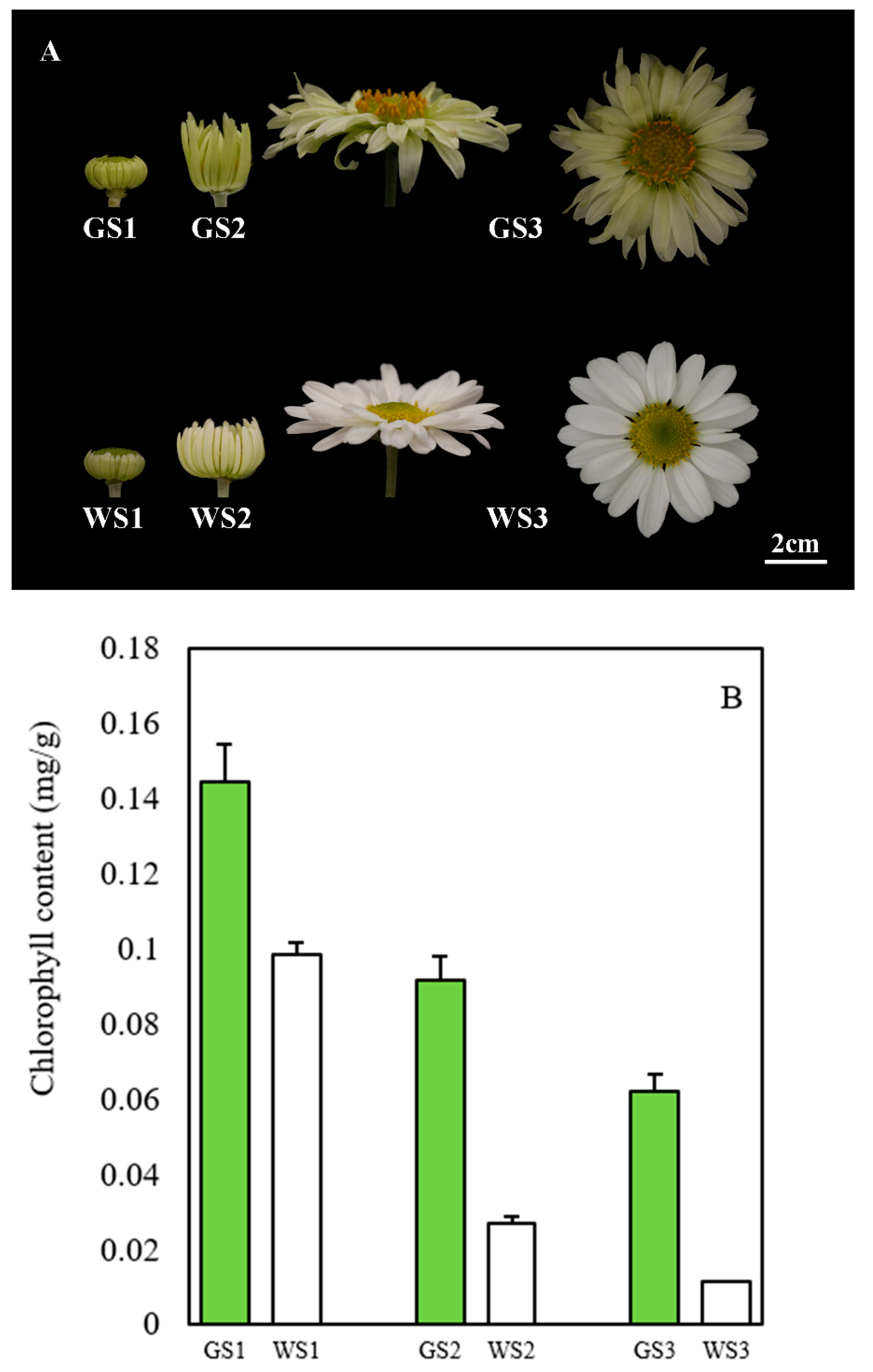
2.2. Measurement of Chlorophyll Content
2.3. Analysis of Plastid Ultrastructure by Transmission Electron Microscopy
2.4. RNA Extraction and Sequencing
2.5. Differential Expression Analysis
2.6. TF Identification
2.7. Weighted Gene Co-Expression Network Analysis
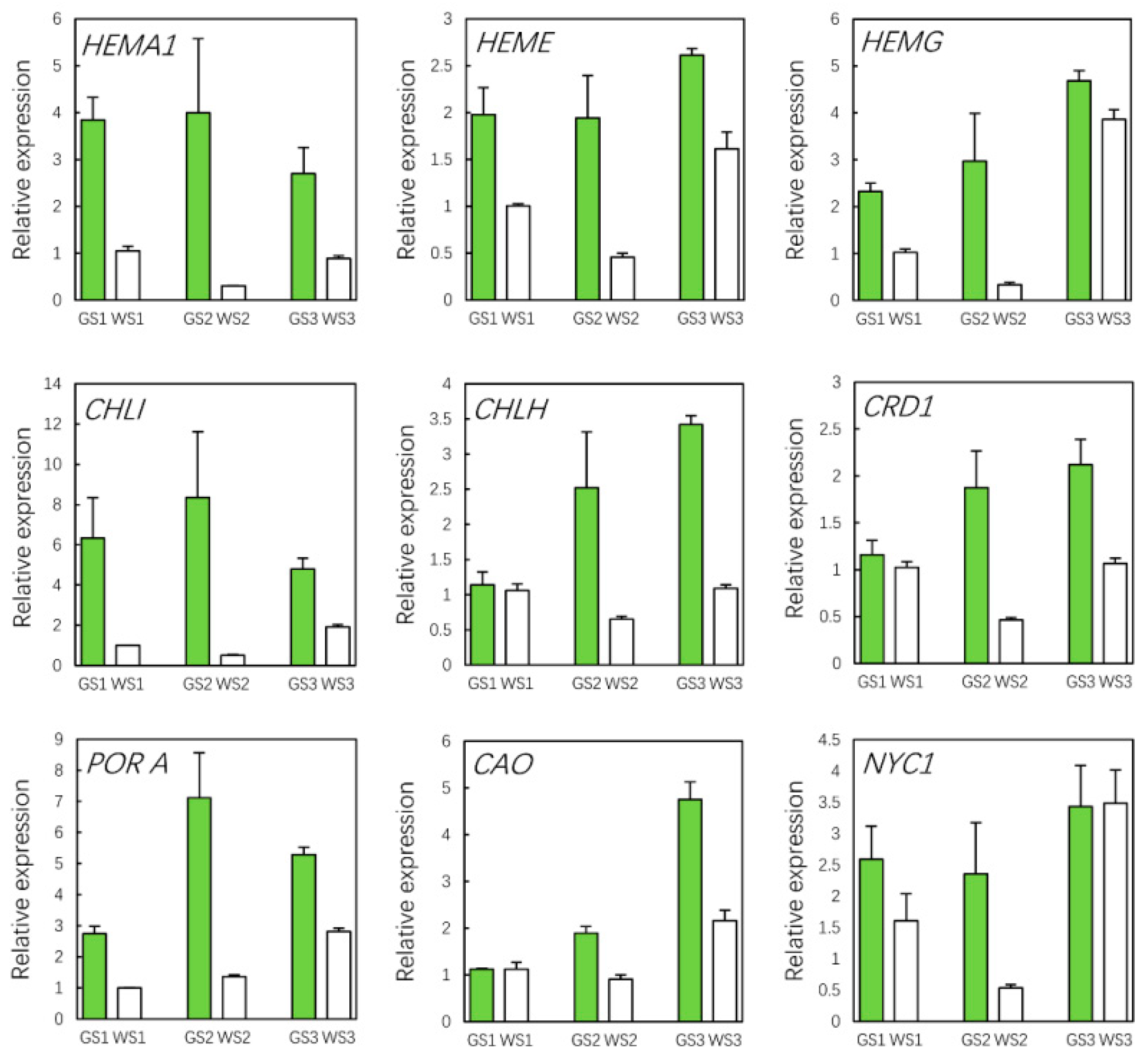
2.8. Quantitative Real-Time-PCR Analysis
3. Results
3.1. Chlorophyll Content of Green and White Ray Florets of Chrysanthemum
3.2. Morphology and Structure of Chloroplasts of Green and White Ray Florets
3.3. RNA-Seq Analysis of Green and White Pools
3.4. Gene Annotation and Functional Classification
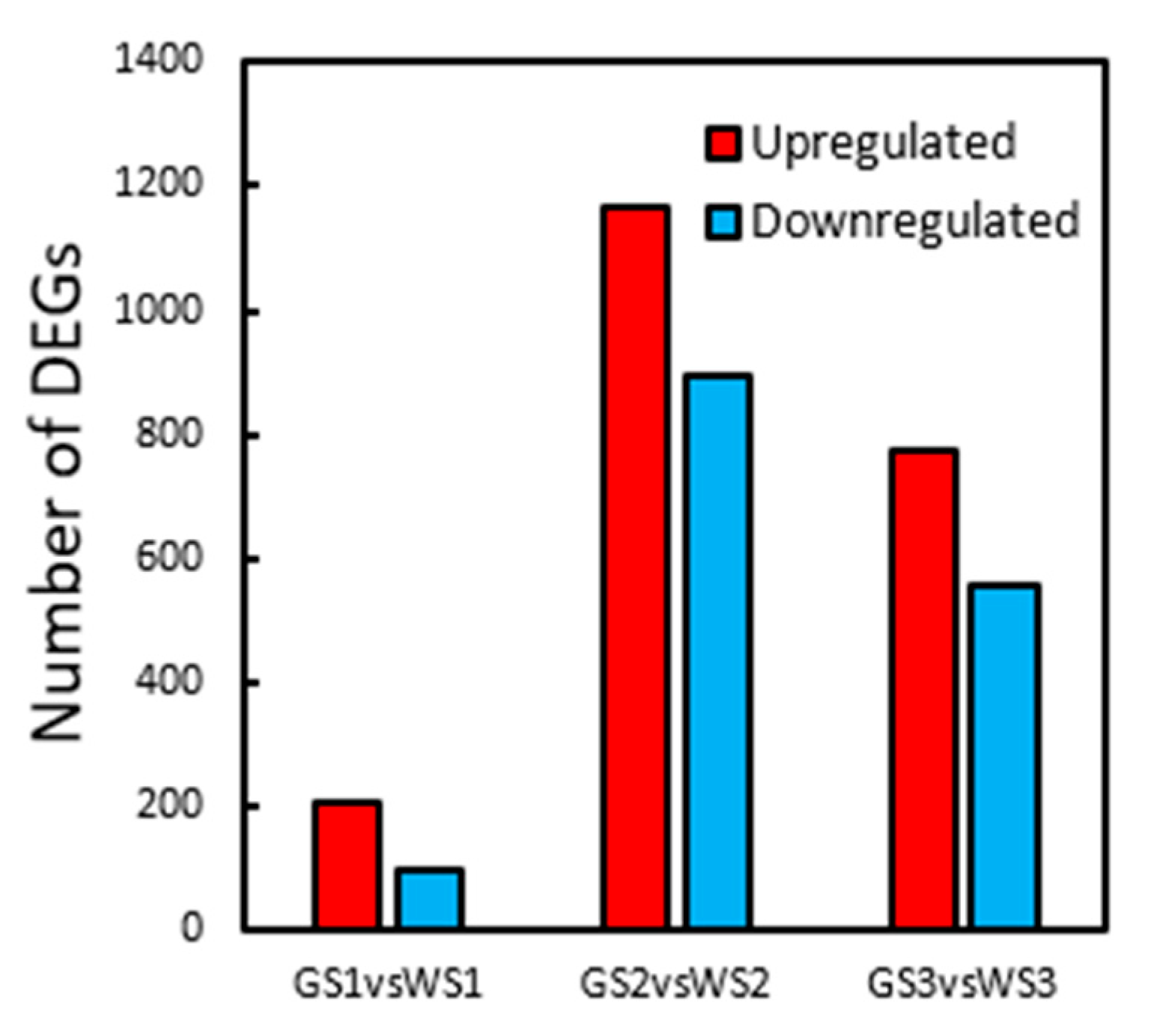
3.5. DEG Analysis
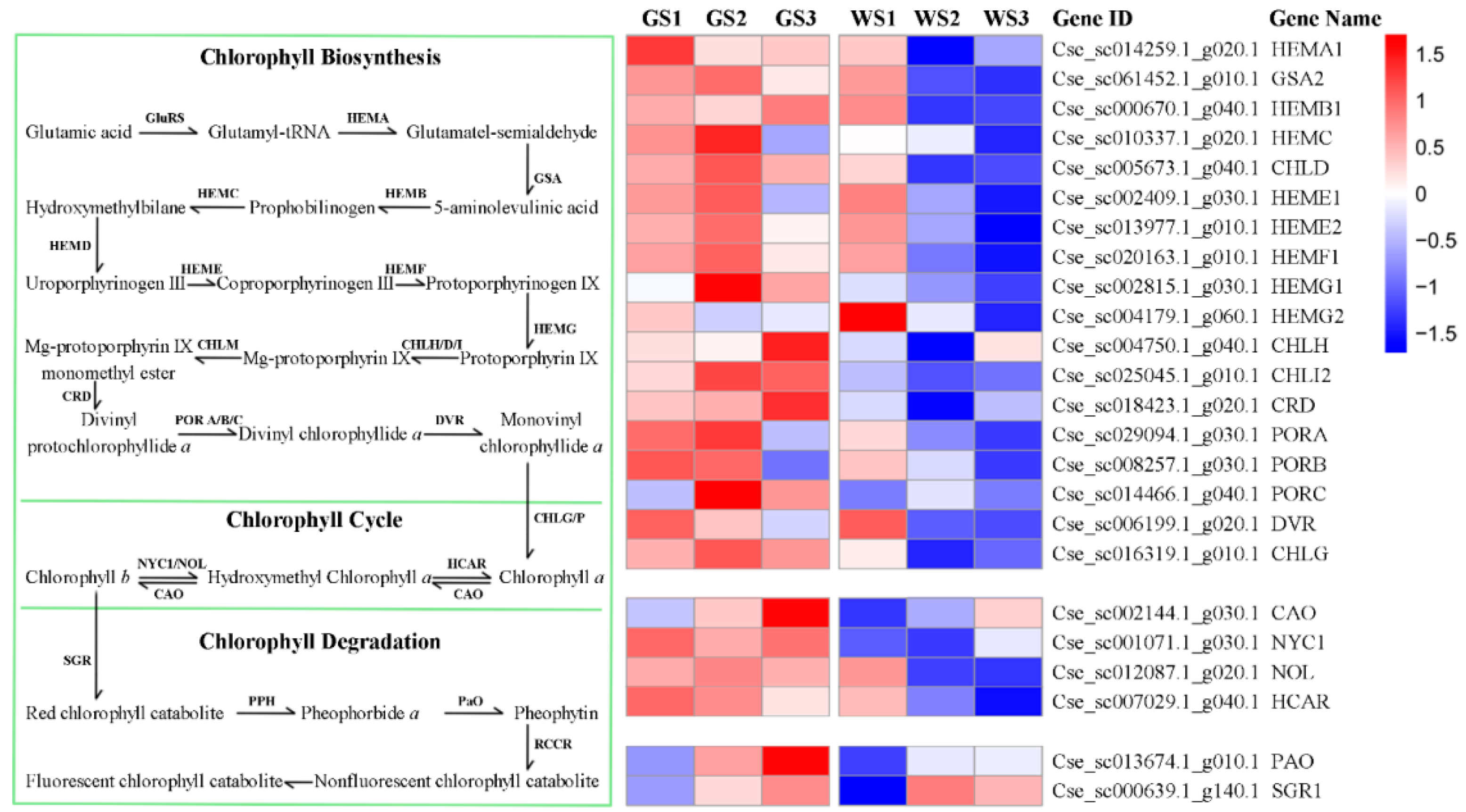
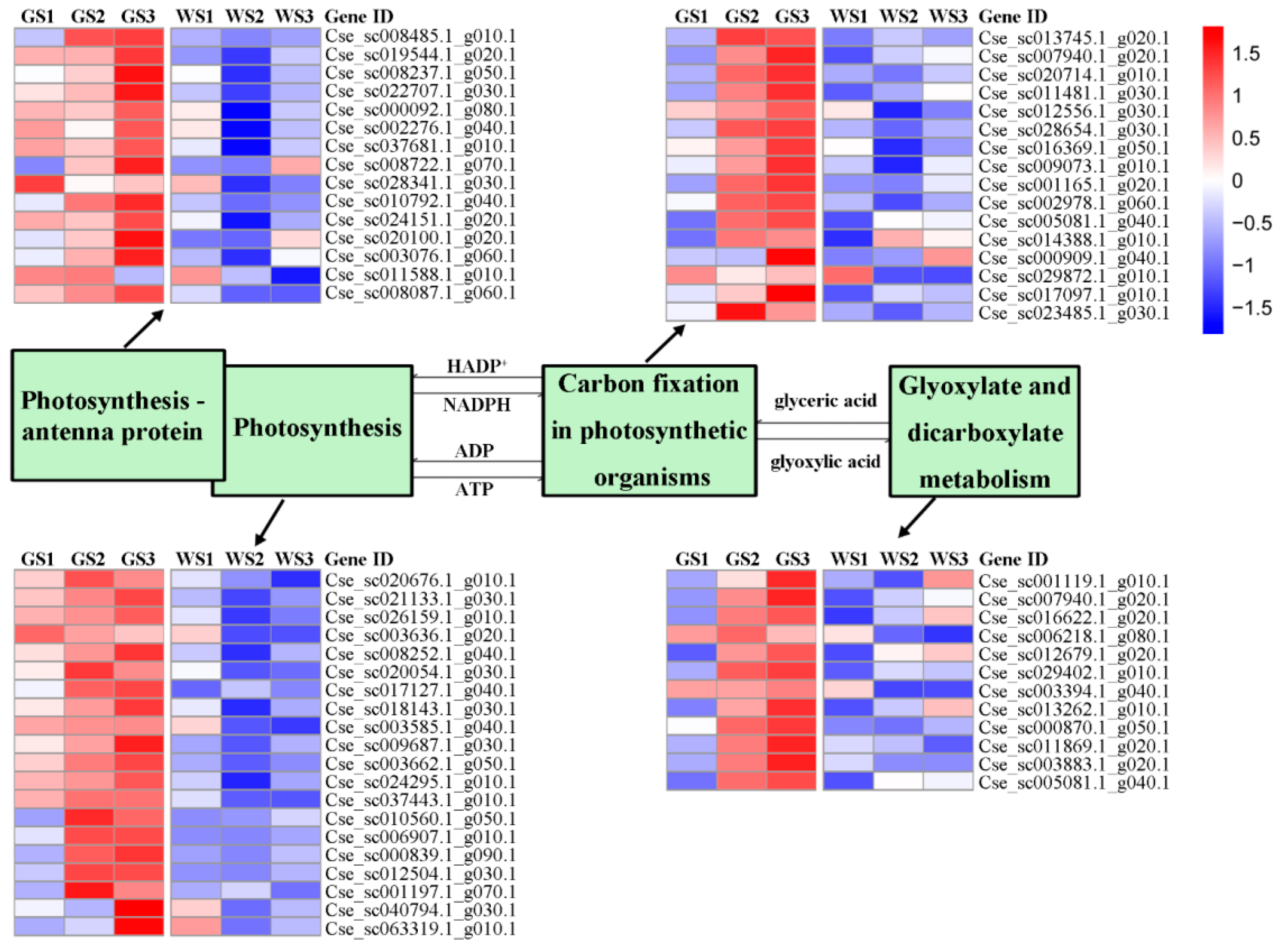
3.6. Expression of Genes Involved in Chlorophyll Metabolism and Related Pathways
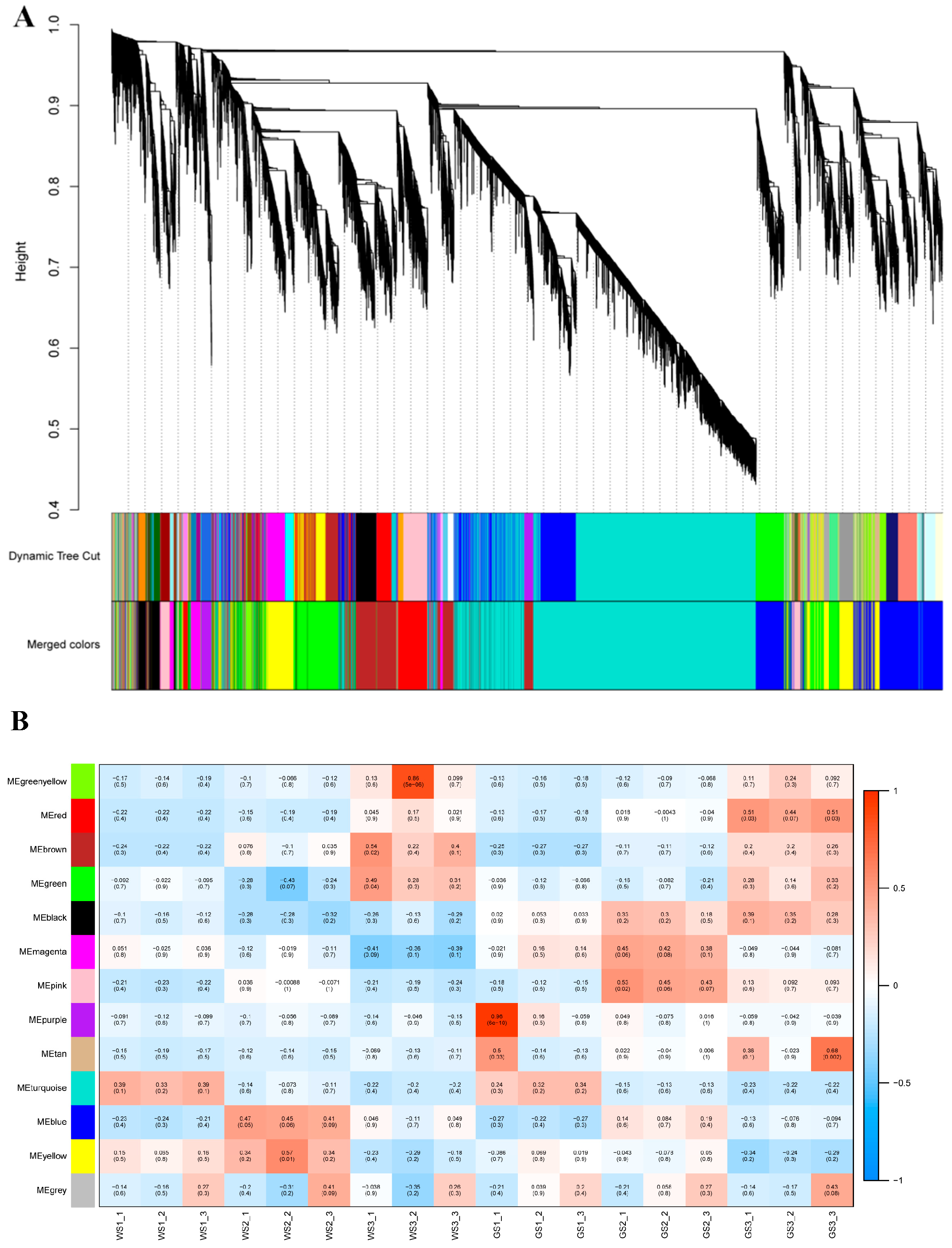
3.7. WGCNA of DEGs
3.8. Functional Enrichment Analysis of Key Modules
3.9. Identification of Hub TFs in the Black Module
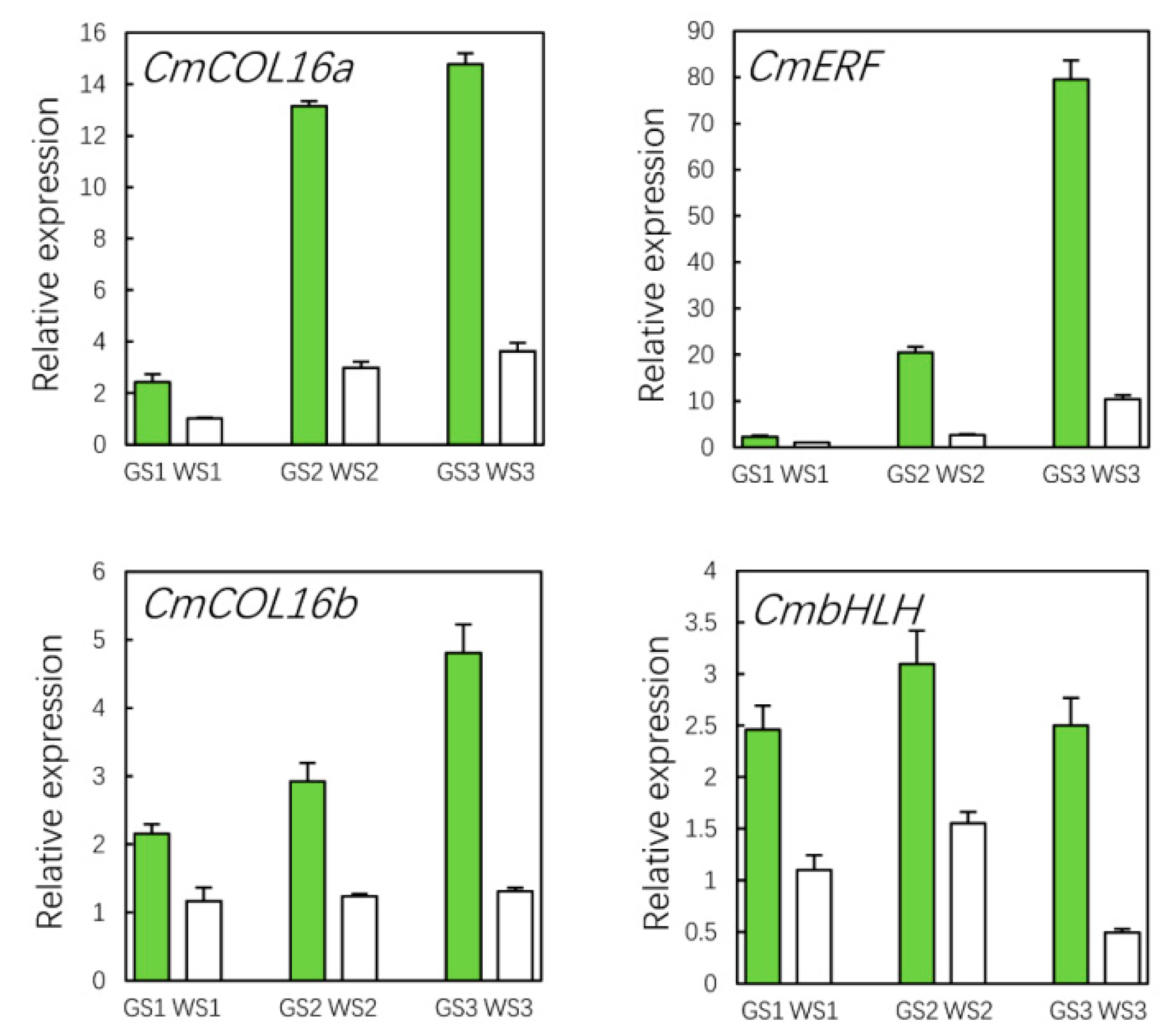
3.10. qRT-PCR Validation of Hub Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, J.Y. The Origin of Garden Chrysanthemum, 1st ed.; Anhui Science and Technology Publishing House: Hefei, China, 2012; pp. 9–11. [Google Scholar]
- Park, C.H.; Chae, S.C.; Park, S.Y.; Kim, J.K.; Kim, Y.J.; Chung, S.O.; Arasu, M.V.; Al-Dhabi, N.A.; Park, S.U. Anthocyanin and carotenoid contents in different cultivars of chrysanthemum (Dendranthema grandiflorum Ramat.) flower. Molecules 2015, 20, 11090–11102. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Liu, X.; Li, H.; Yin, X.; Grierson, D.; Li, F.; Chen, K. CmMYB#7, an R3 MYB transcription factor, acts as a negative regulator of anthocyanin biosynthesis in chrysanthemum. J. Exp. Bot. 2019, 70, 3111–3123. [Google Scholar] [PubMed]
- Noda, N.; Yoshioka, S.; Kishimoto, S.; Nakayama, M.; Douzono, M.; Tanaka, Y.; Aida, R. Generation of blue chrysanthemums by anthocyanin B-ring hydroxylation and glucosylation and its coloration mechanism. Sci. Adv. 2017, 3, e1602785. [Google Scholar] [CrossRef]
- Hörtensteiner, S. Stay-green regulates chlorophyll and chlorophyll-binding protein degradation during senescence. Trends Plant Sci. 2009, 14, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Gao, J.; Zhu, X.; Song, Y.; Li, Z.; Ren, G.; Zhou, X.; Kuai, B. ABF2, ABF3 and ABF4 promote ABA-mediated chlorophyll degradation and leaf senescence by transcriptional activation of chlorophyll catabolic genes and senescence-associated genes in Arabidopsis. Mol. Plant 2016, 9, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Ohmiya, A.; Hirashima, M.; Yagi, M.; Tanase, K.; Yamamizo, C. Identification of genes associated with chlorophyll accumulation in flower petals. PLoS ONE 2014, 9, e113738. [Google Scholar] [CrossRef]
- Ohmiya, A. Molecular mechanisms underlying the diverse array of petal colors in chrysanthemum flowers. Breed Sci. 2018, 68, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A. Investigation on Mechanism of Green Disk Florets of Spray Cut Chrysanthemum. Master of Science Thesis, Nanjing Agricultural University, Nanjing, China, 2014. [Google Scholar]
- Masuda, T.; Fujita, Y. Regulation and evolution of chlorophyll metabolism. Photochem. Photobiol. Sci. 2008, 7, 1131–1149. [Google Scholar] [CrossRef]
- Eckhardt, U.; Grimm, B.; Hörtensteiner, S. Recent advances in chlorophyll biosynthesis and breakdown in higher plants. Plant Mol. Biol. 2004, 56, 1–14. [Google Scholar] [CrossRef]
- Hörtensteiner, S. Update on the biochemistry of chlorophyll breakdown. Plant Mol. Biol. 2013, 82, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, P.; Feng, Y.; Xu, H.; Cao, Y.; Tang, Y.; Yuan, S.; Liu, X.; Ming, J. Spatiotemporal transcriptome analysis provides insights into bicolor tepal development in Lilium “Tiny Padhye”. Front. Plant Sci. 2017, 8, 398. [Google Scholar] [CrossRef]
- Ohmiya, A.; Sasaki, K.; Nashima, K.; Oda-Yamamizo, C.; Hirashima, M.; Sumitomo, K. Transcriptome analysis in petals and leaves of chrysanthemums with different chlorophyll levels. BMC Plant Biol. 2017, 17, 202. [Google Scholar] [CrossRef]
- Tan, X.L.; Fan, Z.Q.; Kuang, J.F.; Lu, W.J.; Reiter, R.J.; Lakshmanan, P.; Su, X.G.; Zhou, J.; Chen, J.Y.; Shan, W. Melatonin delays leaf senescence of Chinese flowering cabbage by suppressing ABFs-mediated abscisic acid biosynthesis and chlorophyll degradation. J. Pineal Res. 2019, 6, e12570. [Google Scholar] [CrossRef]
- Li, S.; Gao, J.; Yao, L.; Ren, G.; Zhu, X.; Gao, S.; Qiu, K.; Zhou, X.; Kuai, B. The role of ANAC072 in the regulation of chlorophyll degradation during age- and dark-induced leaf senescence. Plant Cell Rep. 2016, 35, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, Y.; Yang, X.; Pan, Q.; Ma, G.; Bao, M.; Zheng, B.; Duanmu, D.; Lin, R.; Larkin, R.M.; et al. Overexpression of particular MADS-Box transcription factors in heat-stressed plants induces chloroplast biogenesis in petals. Plant Cell Environ. 2019, 42, 1545–1560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Xie, H.; Wei, X.; Dossa, K.; Yu, Y.; Hui, S.; Tang, G.; Zeng, X.; Yu, Y.; Hu, P.; et al. WGCNA analysis of salt-responsive core transcriptome identifies novel hub genes in rice. Genes 2019, 10, 719. [Google Scholar] [CrossRef]
- Huang, D.; Zhao, Y.; Cao, M.; Qiao, L.; Zheng, Z.L. Integrated systems biology analysis of transcriptomes reveals candidate genes for acidity control in developing fruits of sweet orange (Citrus sinensis L. Osbeck). Front. Plant Sci. 2016, 7, 486. [Google Scholar] [CrossRef]
- Bai, Y.; Dougherty, L.; Cheng, L.; Zhong, G.Y.; Xu, K. Uncovering co-expression gene network modules regulating fruit acidity in diverse apples. BMC Genom. 2015, 16, 612. [Google Scholar] [CrossRef]
- Sun, B.; Zhou, X.; Chen, C.; Chen, C.; Chen, K.; Chen, M.; Liu, S.; Chen, G.; Cao, B.; Cao, F.; et al. Coexpression network analysis reveals an MYB transcriptional activator involved in capsaicinoid biosynthesis in hot peppers. Hortic. Res. 2020, 7, 162. [Google Scholar] [CrossRef]
- Liu, Y.; Xin, J.; Liu, L.; Song, A.; Guan, Z.; Fang, W.; Chen, F. A temporal gene expression map of Chrysanthemum leaves infected with Alternaria alternata reveals different stages of defense mechanisms. Hortic. Res. 2020, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Pu, Y.; Liu, Y.; Li, Y.; Qu, J.; Huang, H.; Dai, S. Comparative transcriptomics and weighted gene co-expression correlation network analysis (WGCNA) reveal potential regulation mechanism of carotenoid accumulation in Chrysanthemum × morifolium. Plant Physiol. Biochem. 2019, 142, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhang, Y.; Zhao, Y.; Luo, T.; Deng, H.; Meng, C.; Wang, C. Heterosis and mixed genetic analysis of flower head traits in the progeny population of a crossing between Chrysanthemum × morifolium ‘Lüdingdang’ and Chrysanthemum vestitum. Acta Hortic. Sinica 2021, 48, 96–106. [Google Scholar]
- Liu, Y.; Roof, S.; Ye, Z.; Barry, C.; van Tuinen, A.; Vrebalov, J.; Bowler, C.; Giovannoni, J. Manipulation of light signal transduction as a means of modifying fruit nutritional quality in tomato. Proc. Natl. Acad. Sci. USA 2004, 101, 9897–9902. [Google Scholar] [CrossRef]
- Hirakawa, H.; Sumitomo, K.; Hisamatsu, T.; Nagano, S.; Shirasawa, K.; Higuchi, Y.; Kusaba, M.; Koshioka, M.; Nakano, Y.; Yagi, M.; et al. De novo whole-genome assembly in Chrysanthemum seticuspe, a model species of Chrysanthemums, and its application to genetic and gene discovery analysis. DNA Res. 2019, 26, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2DDCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Shim, M.S.; Sim, S.-C.; Oh, K.H.; Seo, J.Y. Genetic variation of flower characteristics in a population derived from a cross between the chrysanthemum cultivars ‘Falcao’ and ‘Frill Green’. Hortic. Environ. Biotechnol. 2014, 55, 322–328. [Google Scholar] [CrossRef]
- Xue, H.; Shi, T.; Wang, F.; Zhou, H.; Yang, J.; Wang, L.; Wang, S.; Su, Y.; Zhang, Z.; Qiao, Y.; et al. Interval mapping for red/green skin color in Asian pears using a modified QTL-seq method. Hortic Res 2017, 4, 17053. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Li, Y.; Ma, C.; Feili Du, L.T.; Xu, M. Combined genome-wide association study and transcriptome analysis reveal candidate genes for resistance to Fusarium ear rot in maize. J. Integr. Plant Biol. 2020, 62, 1535–1551. [Google Scholar] [CrossRef]
- Van Geest, G.; Bourke, P.M.; Voorrips, R.E.; Marasek-Ciolakowska, A.; Liao, Y.; Post, A.; van Meeteren, U.; Visser, R.; Maliepaard, C.; Arens, P. An ultra-dense integrated linkage map for hexaploid chrysanthemum enables multi-allelic QTL analysis. Theor. Appl. Genet. 2017, 12, 2527–2541. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef]
- Edae, E.A.; Rouse, M.N. Bulked segregant analysis RNA-seq (BSR-Seq) validated a stem resistance locus in Aegilops umbellulata, a wild relative of wheat. PLoS ONE 2019, 14, e0215492. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Zheng, F.; Wang, K.; Xu, Y.; Xu, H.; Shen, W.; Zhu, C.; Zhang, X.; Sui, W.; Tang, D.; et al. A single-cell map for the transcriptomic signatures of peripheral blood mononuclear cells in end-stage renal disease. Nephrol. Dial. Transplant. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhao, K.; Zhang, X.; Song, A.; Su, J.; Hu, Y.; Zhao, W.; Jiang, J.; Chen, F. Comprehensive characterization of a floral mutant reveals the mechanism of hooked petal morphogenesis in Chrysanthemum Morifolium. Plant Biotechnol. J. 2019, 17, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, S.; Dunford, R.P.; Coupland, G.; Laurie, D.A. The Evolution of CONSTANS-like gene families in barley, rice, and Arabidopsis. Plant Physiol. 2003, 131, 1855–1867. [Google Scholar] [CrossRef] [PubMed]
- Ohmiya, A.; Oda-Yamamizo, C.; Kishimoto, S. Overexpression of CONSTANS-like 16 enhances chlorophyll accumulation in petunia corollas. Plant Sci. 2019, 280, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Licausi, F.; Ohme-Takagi, M.; Perata, P. APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: Mediators of stress responses and developmental programs. New Phytol. 2013, 199, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xie, X.; Liu, S.; Chen, K.; Yin, X. Auto- and mutual-regulation between two CitERFs contribute to ethylene-induced citrus fruit degreening. Food Chem. 2019, 299, 125163. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Y.; Jiao, Y.; Wu, J.; Zhang, Z.; Yu, X.; Ma, Y. Transcriptome profiling of yellow leafy head development during the heading stage in Chinese cabbage (Brassica rapa subsp. pekinensis). Physiol. Plant 2018, 165, 800–813. [Google Scholar] [CrossRef]
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Qiu, J.Y.; Hui, Q.L.; Xu, Y.Y.; He, Y.Z.; Peng, L.Z.; Fu, X.Z. Systematic analysis of the basic/helix-loop-helix (bHLH) transcription factor family in pummelo (Citrus grandis) and identification of the key members involved in the response to iron deficiency. BMC Genom. 2020, 21, 233. [Google Scholar] [CrossRef]
- Kiełbowicz-Matuk, A. Involvement of plant C2H2-type zinc finger transcription factors in stress responses. Plant Sci. 2012, 185–186, 78–85. [Google Scholar] [CrossRef]
- Salih, H.; Odongo, M.R.; Gong, W.; He, S.; Du, X. Genome-wide analysis of cotton C2H2-zinc finger transcription factor family and their expression analysis during fiber development. BMC Plant Biol. 2019, 19, 400. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Library | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|---|---|
| GS1_1 | FRAS192310180-1r | 69,909,924 | 67,974,534 | 10.2G | 0.02 | 98.22 | 94.50 | 41.89 |
| GS1_2 | FRAS192310181-1r | 67,154,326 | 65,518,128 | 9.83G | 0.02 | 98.28 | 94.64 | 42.19 |
| GS1_3 | FRAS192310182-1r | 79,385,102 | 77,582,694 | 11.64G | 0.02 | 98.16 | 94.41 | 42.38 |
| WS1_1 | FRAS192310183-1r | 78,199,758 | 76,158,198 | 11.42G | 0.02 | 98.26 | 94.69 | 42.45 |
| WS1_2 | FRAS192310184-1r | 68,968,074 | 67,390,906 | 10.11G | 0.02 | 98.25 | 94.55 | 42.33 |
| WS1_3 | FRAS192310185-1r | 71,120,720 | 69,206,430 | 10.38G | 0.02 | 98.10 | 94.24 | 42.40 |
| GS2_1 | FRAS192310186-1r | 74,730,896 | 72,493,234 | 10.87G | 0.02 | 98.19 | 94.48 | 42.38 |
| GS2_2 | FRAS192310187-1r | 72,994,076 | 70,710,496 | 10.61G | 0.02 | 98.25 | 94.63 | 42.26 |
| GS2_3 | FRAS192310188-1r | 73,085,728 | 71,432,126 | 10.71G | 0.02 | 98.11 | 94.23 | 42.16 |
| WS2_1 | FRAS192310189-1r | 68,345,294 | 66,024,344 | 9.9G | 0.02 | 98.16 | 94.45 | 41.92 |
| WS2_2 | FRAS192310190-1r | 81,521,172 | 79,358,278 | 11.9G | 0.02 | 98.19 | 94.56 | 41.69 |
| WS2_3 | FRAS192310191-1r | 69,440,584 | 67,776,290 | 10.17G | 0.02 | 98.08 | 94.24 | 41.79 |
| GS3_1 | FRAS192042324-1r | 76,123,912 | 74,489,046 | 11.17G | 0.03 | 97.90 | 93.72 | 41.99 |
| GS3_2 | FRAS192042325-1r | 76,239,258 | 74,539,470 | 11.18G | 0.03 | 98.00 | 93.99 | 42.07 |
| GS3_3 | FRAS192042326-1r | 75,168,204 | 73,433,218 | 11.01G | 0.03 | 97.92 | 93.74 | 42.05 |
| WS3_1 | FRAS192042327-1r | 78,303,734 | 76,184,148 | 11.43G | 0.03 | 97.84 | 93.61 | 41.70 |
| WS3_2 | FRAS192042328-1r | 78,068,928 | 76,158,448 | 11.42G | 0.03 | 97.92 | 93.77 | 41.68 |
| WS3_3 | FRAS192042329-1r | 79,621,212 | 77,866,790 | 11.68G | 0.03 | 97.82 | 93.55 | 41.99 |
| KEGG Pathway | No. in All Unigenes | No. in DEGs |
|---|---|---|
| Photosynthesis | 151 | 20 |
| Photosynthesis—antenna proteins | 65 | 15 |
| Chlorophyll metabolism | 113 | 24 |
| Carbon fixation in photosynthetic organisms | 230 | 16 |
| Glyoxylate and dicarboxylate metabolism | 204 | 12 |
| Module | Number of DEGs Related to Key Processes in Chrysanthemum | |||||
|---|---|---|---|---|---|---|
| Total DEGs | Photosynthesis—Antenna Proteins | Photosynthesis | Carbon Fixation in Photosynthetic Organisms | Glyoxylate and Dicarboxylate Metabolism | Chlorophyll Metabolism | |
| Red | 116 | 5 | 0 | 0 | 0 | 0 |
| Black | 97 | 6 | 20 | 10 | 7 | 5 |
| Other | 1621 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, H.; Zeng, T.; Zhao, Y.; Luo, T.; Deng, H.; Meng, C.; Luo, J.; Wang, C. Identification of Chlorophyll Metabolism- and Photosynthesis-Related Genes Regulating Green Flower Color in Chrysanthemum by Integrative Transcriptome and Weighted Correlation Network Analyses. Genes 2021, 12, 449. https://doi.org/10.3390/genes12030449
Fu H, Zeng T, Zhao Y, Luo T, Deng H, Meng C, Luo J, Wang C. Identification of Chlorophyll Metabolism- and Photosynthesis-Related Genes Regulating Green Flower Color in Chrysanthemum by Integrative Transcriptome and Weighted Correlation Network Analyses. Genes. 2021; 12(3):449. https://doi.org/10.3390/genes12030449
Chicago/Turabian StyleFu, Hansen, Tuo Zeng, Yangyang Zhao, Tingting Luo, Huijie Deng, Chenwei Meng, Jing Luo, and Caiyun Wang. 2021. "Identification of Chlorophyll Metabolism- and Photosynthesis-Related Genes Regulating Green Flower Color in Chrysanthemum by Integrative Transcriptome and Weighted Correlation Network Analyses" Genes 12, no. 3: 449. https://doi.org/10.3390/genes12030449
APA StyleFu, H., Zeng, T., Zhao, Y., Luo, T., Deng, H., Meng, C., Luo, J., & Wang, C. (2021). Identification of Chlorophyll Metabolism- and Photosynthesis-Related Genes Regulating Green Flower Color in Chrysanthemum by Integrative Transcriptome and Weighted Correlation Network Analyses. Genes, 12(3), 449. https://doi.org/10.3390/genes12030449