
The Amazing Acrobat: Yeast’s Histone H3K56 Juggles Several Important Roles While Maintaining Perfect Balance


Abstract
1. Introduction
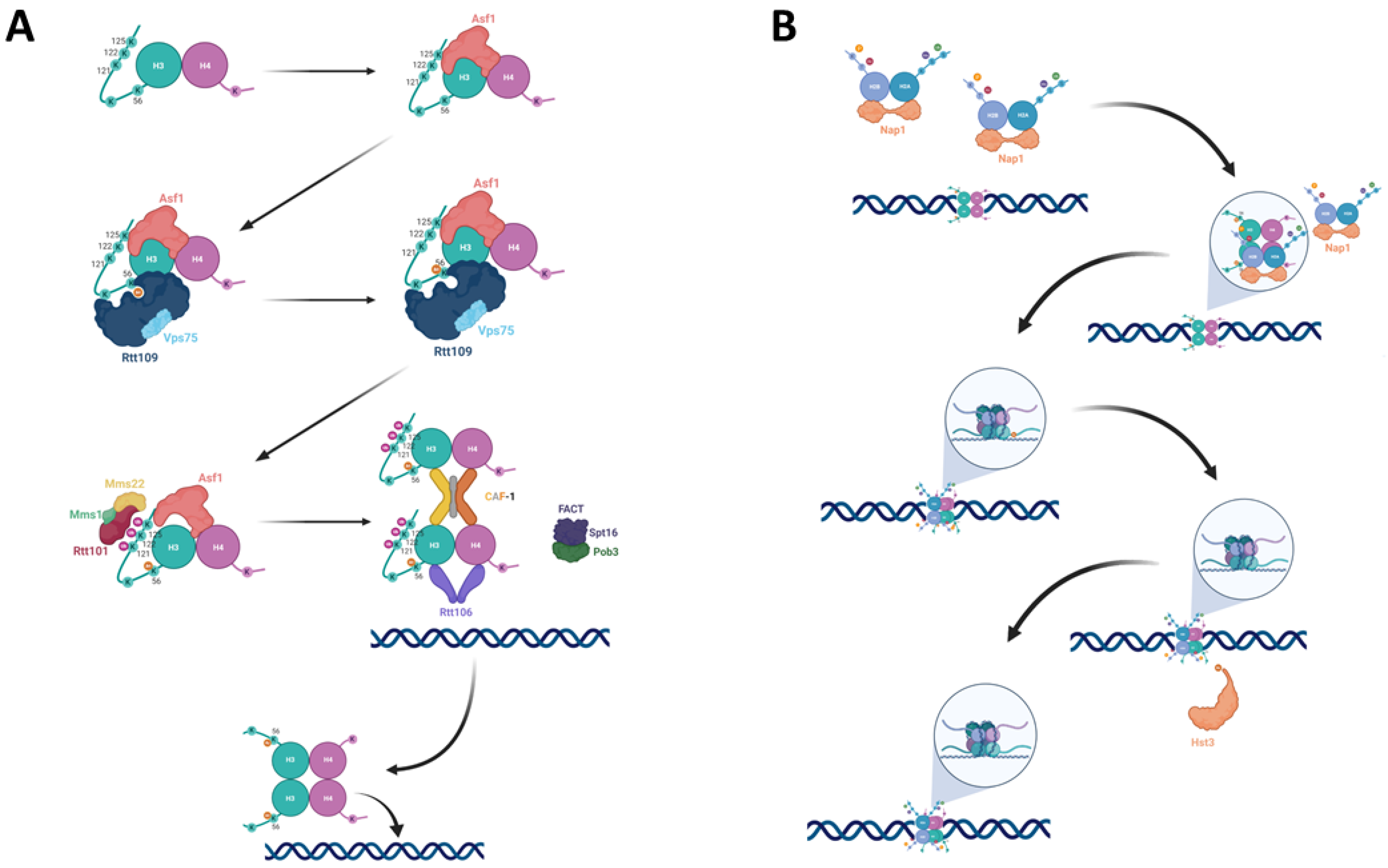
2. From Acetylation to Incorporation
2.1. Asf1
2.2. Rtt109
2.3. Rtt101
2.4. Chromatin Remodelers
2.5. Nucleosome Assembly
3. Histone Deacetylation
3.1. Hst3
3.2. Hst4
3.3. Δhst3 Δhst4
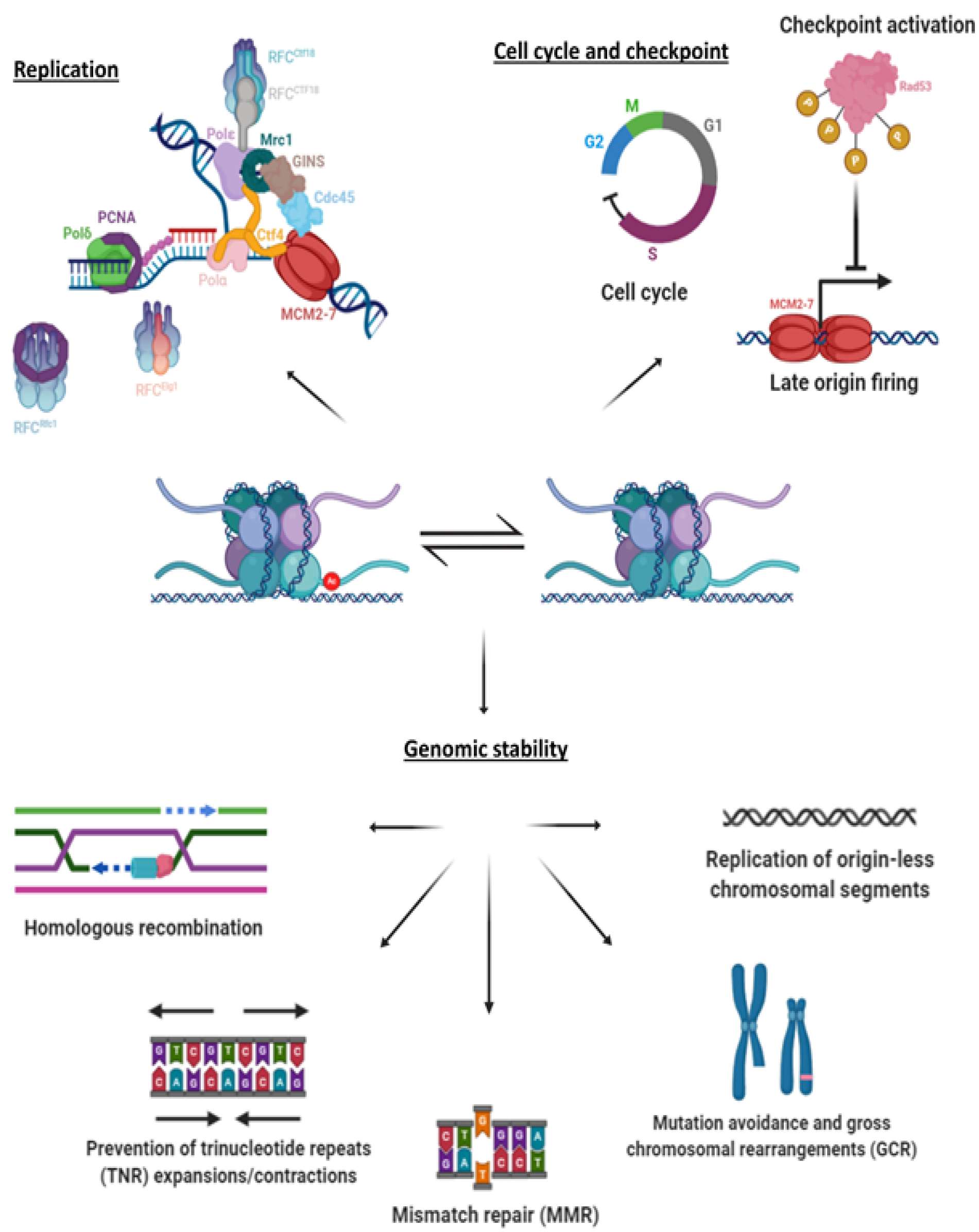
4. Hyper-Acetylation and Replication
5. Hyper-acetylation and Genome Stability
5.1. Checkpoint Activation and Recovery
5.2. Mutations and Genomic Instability
5.3. Repair Pathways
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cutter, A.R.; Hayes, J.J. A brief review of nucleosome structure. FEBS Lett. 2015, 589, 2914–2922. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Fillingham, J.; Recht, J.; Silva, A.C.; Suter, B.; Emili, A.; Stagljar, I.; Krogan, N.J.; Allis, C.D.; Keogh, M.C.; Greenblatt, J.F. Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol. Cell. Biol. 2008, 28, 4342–4353. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Lucca, C.; Vanoli, F.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Haber, J.; Foiani, M. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 2004, 23, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, R.; Hudson, A.; Jackson, S.P. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 2007, 315, 649–652. [Google Scholar] [CrossRef]
- Han, J.; Zhou, H.; Horazdovsky, B.; Zhang, K.; Xu, R.M.; Zhang, Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 2007, 315, 653–655. [Google Scholar] [CrossRef]
- Rufiange, A.; Jacques, P.E.; Bhat, W.; Robert, F.; Nourani, A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol. Cell 2007, 27, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Laniel, M.A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Neumann, H.; Hancock, S.M.; Buning, R.; Routh, A.; Chapman, L.; Somers, J.; Owen-Hughes, T.; van Noort, J.; Rhodes, D.; Chin, J.W. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 2009, 36, 153–163. [Google Scholar] [CrossRef]
- Ferreira, H.; Somers, J.; Webster, R.; Flaus, A.; Owen-Hughes, T. Histone tails and the H3 alphaN helix regulate nucleosome mobility and stability. Mol. Cell. Biol. 2007, 27, 4037–4048. [Google Scholar] [CrossRef]
- Masumoto, H.; Hawke, D.; Kobayashi, R.; Verreault, A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 2005, 436, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Parthun, M.R. The nuclear Hat1p/Hat2p complex: A molecular link between type B histone acetyltransferases and chromatin assembly. Mol. Cell 2004, 14, 195–205. [Google Scholar] [CrossRef]
- Verreault, A.; Kaufman, P.D.; Kobayashi, R.; Stillman, B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell 1996, 87, 95–104. [Google Scholar] [CrossRef]
- Hyland, E.M.; Cosgrove, M.S.; Molina, H.; Wang, D.; Pandey, A.; Cottee, R.J.; Boeke, J.D. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol. Cell. Biol. 2005, 25, 10060–10070. [Google Scholar] [CrossRef] [PubMed]
- Adkins, M.W.; Howar, S.R.; Tyler, J.K. Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol. Cell 2004, 14, 657–666. [Google Scholar] [CrossRef]
- Schwabish, M.A.; Struhl, K. Asf1 mediates histone eviction and deposition during elongation by RNA polymerase II. Mol. Cell 2006, 22, 415–422. [Google Scholar] [CrossRef]
- English, C.M.; Adkins, M.W.; Carson, J.J.; Churchill, M.E.; Tyler, J.K. Structural basis for the histone chaperone activity of Asf1. Cell 2006, 127, 495–508. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Henry, R.A.; Huang, L.; Chen, X.; Stargell, L.A.; Andrews, A.J. Utilizing targeted mass spectrometry to demonstrate Asf1-dependent increases in residue specificity for Rtt109-Vps75 mediated histone acetylation. PLoS ONE 2015, 10, e0118516. [Google Scholar] [CrossRef]
- Chen, S.H.; Albuquerque, C.P.; Liang, J.; Suhandynata, R.T.; Zhou, H. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J. Biol. Chem. 2010, 285, 12803–12812. [Google Scholar] [CrossRef]
- Berndsen, C.E.; Tsubota, T.; Lindner, S.E.; Lee, S.; Holton, J.M.; Kaufman, P.D.; Keck, J.L.; Denu, J.M. Molecular functions of the histone acetyltransferase chaperone complex Rtt109-Vps75. Nat. Struct. Mol. Biol. 2008, 15, 948–956. [Google Scholar] [CrossRef]
- Park, Y.J.; Sudhoff, K.B.; Andrews, A.J.; Stargell, L.A.; Luger, K. Histone chaperone specificity in Rtt109 activation. Nat. Struct. Mol. Biol. 2008, 15, 957–964. [Google Scholar] [CrossRef]
- Albaugh, B.N.; Arnold, K.M.; Lee, S.; Denu, J.M. Autoacetylation of the histone acetyltransferase Rtt109. J. Biol. Chem. 2011, 286, 24694–24701. [Google Scholar] [CrossRef]
- Zhang, L.; Serra-Cardona, A.; Zhou, H.; Wang, M.; Yang, N.; Zhang, Z.; Xu, R.M. Multisite Substrate Recognition in Asf1-Dependent Acetylation of Histone H3 K56 by Rtt109. Cell 2018, 174, 818–830.e1. [Google Scholar] [CrossRef]
- Alcasabas, A.A.; Osborn, A.J.; Bachant, J.; Hu, F.; Werler, P.J.; Bousset, K.; Furuya, K.; Diffley, J.F.; Carr, A.M.; Elledge, S.J. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 2001, 3, 958–965. [Google Scholar] [CrossRef]
- Donham, D.C., 2nd; Scorgie, J.K.; Churchill, M.E. The activity of the histone chaperone yeast Asf1 in the assembly and disassembly of histone H3/H4-DNA complexes. Nucleic Acids Res. 2011, 39, 5449–5458. [Google Scholar] [CrossRef][Green Version]
- Han, J.; Li, Q.; McCullough, L.; Kettelkamp, C.; Formosa, T.; Zhang, Z. Ubiquitylation of FACT by the cullin-E3 ligase Rtt101 connects FACT to DNA replication. Genes Dev. 2010, 24, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, I.W.; Rabut, G.; Poveda, A.; Scheel, H.; Malmstrom, J.; Ulrich, H.; Hofmann, K.; Pasero, P.; Peter, M.; Luke, B. Rtt101 and Mms1 in budding yeast form a CUL4(DDB1)-like ubiquitin ligase that promotes replication through damaged DNA. EMBO Rep. 2008, 9, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.R.; Miller, K.M.; Maas, N.L.; Roguev, A.; Fillingham, J.; Chu, C.S.; Schuldiner, M.; Gebbia, M.; Recht, J.; Shales, M.; et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 2007, 446, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.D.; Zhou, H.; Dar, M.A.; Zhang, Z.; Luger, K. Yeast CAF-1 assembles histone (H3-H4)2 tetramers prior to DNA deposition. Nucleic Acids Res. 2012, 40, 10139–10149. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Hu, Q.; Li, Q.; Thompson, J.R.; Cui, G.; Fazly, A.; Davies, B.A.; Botuyan, M.V.; Zhang, Z.; Mer, G. Structural basis for recognition of H3K56-acetylated histone H3-H4 by the chaperone Rtt106. Nature 2012, 483, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, H.; Wurtele, H.; Davies, B.; Horazdovsky, B.; Verreault, A.; Zhang, Z. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 2008, 134, 244–255. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Feng, J.; Leng, H.; Li, S.; Xiao, J.; Liu, S.; Xu, Z.; Xu, J.; Li, D.; et al. The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Rep. 2016, 14, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Bulger, M.; Muramatsu, M.; Ito, T. Multistep chromatin assembly on supercoiled plasmid DNA by nucleosome assembly protein-1 and ATP-utilizing chromatin assembly and remodeling factor. J. Biol. Chem. 2001, 276, 27384–27391. [Google Scholar] [CrossRef] [PubMed]
- Akey, C.W.; Luger, K. Histone chaperones and nucleosome assembly. Curr. Opin. Struct. Biol. 2003, 13, 6–14. [Google Scholar] [CrossRef]
- Groth, A.; Rocha, W.; Verreault, A.; Almouzni, G. Chromatin challenges during DNA replication and repair. Cell 2007, 128, 721–733. [Google Scholar] [CrossRef]
- Groth, A.; Corpet, A.; Cook, A.J.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of replication fork progression through histone supply and demand. Science 2007, 318, 1928–1931. [Google Scholar] [CrossRef]
- Yu, C.; Gan, H.; Serra-Cardona, A.; Zhang, L.; Gan, S.; Sharma, S.; Johansson, E.; Chabes, A.; Xu, R.M.; Zhang, Z. A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 2018, 361, 1386–1389. [Google Scholar] [CrossRef]
- Brachmann, C.B.; Sherman, J.M.; Devine, S.E.; Cameron, E.E.; Pillus, L.; Boeke, J.D. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995, 9, 2888–2902. [Google Scholar] [CrossRef]
- Celic, I.; Masumoto, H.; Griffith, W.P.; Meluh, P.; Cotter, R.J.; Boeke, J.D.; Verreault, A. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr. Biol. 2006, 16, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Maas, N.L.; Miller, K.M.; DeFazio, L.G.; Toczyski, D.P. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol. Cell 2006, 23, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Spellman, P.T.; Volpe, T.; Brown, P.O.; Botstein, D.; Davis, T.N.; Futcher, B. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 2000, 406, 90–94. [Google Scholar] [CrossRef]
- Delgoshaie, N.; Tang, X.; Kanshin, E.D.; Williams, E.C.; Rudner, A.D.; Thibault, P.; Tyers, M.; Verreault, A. Regulation of the histone deacetylase Hst3 by cyclin-dependent kinases and the ubiquitin ligase SCFCdc4. J. Biol. Chem. 2014, 289, 13186–13196. [Google Scholar] [CrossRef]
- Ozdemir, A.; Spicuglia, S.; Lasonder, E.; Vermeulen, M.; Campsteijn, C.; Stunnenberg, H.G.; Logie, C. Characterization of lysine 56 of histone H3 as an acetylation site in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 25949–25952. [Google Scholar] [CrossRef] [PubMed]
- Sia, R.A.; Herald, H.A.; Lew, D.J. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol. Biol. Cell 1996, 7, 1657–1666. [Google Scholar] [CrossRef][Green Version]
- Edenberg, E.R.; Vashisht, A.A.; Topacio, B.R.; Wohlschlegel, J.A.; Toczyski, D.P. Hst3 is turned over by a replication stress-responsive SCF(Cdc4) phospho-degron. Proc. Natl. Acad. Sci. USA 2014, 111, 5962–5967. [Google Scholar] [CrossRef] [PubMed]
- Thaminy, S.; Newcomb, B.; Kim, J.; Gatbonton, T.; Foss, E.; Simon, J.; Bedalov, A. Hst3 is regulated by Mec1-dependent proteolysis and controls the S phase checkpoint and sister chromatid cohesion by deacetylating histone H3 at lysine 56. J. Biol. Chem. 2007, 282, 37805–37814. [Google Scholar] [CrossRef]
- Irene, C.; Theis, J.F.; Gresham, D.; Soteropoulos, P.; Newlon, C.S. Hst3p, a histone deacetylase, promotes maintenance of Saccharomyces cerevisiae chromosome III lacking efficient replication origins. Mol. Genet. Genom. MGG 2016, 291, 271–283. [Google Scholar] [CrossRef]
- Madsen, C.T.; Sylvestersen, K.B.; Young, C.; Larsen, S.C.; Poulsen, J.W.; Andersen, M.A.; Palmqvist, E.A.; Hey-Mogensen, M.; Jensen, P.B.; Treebak, J.T.; et al. Biotin starvation causes mitochondrial protein hyperacetylation and partial rescue by the SIRT3-like deacetylase Hst4p. Nat. Commun. 2015, 6, 7726. [Google Scholar] [CrossRef]
- Simoneau, A.; Ricard, E.; Weber, S.; Hammond-Martel, I.; Wong, L.H.; Sellam, A.; Giaever, G.; Nislow, C.; Raymond, M.; Wurtele, H. Chromosome-wide histone deacetylation by sirtuins prevents hyperactivation of DNA damage-induced signaling upon replicative stress. Nucleic Acids Res. 2016, 44, 2706–2726. [Google Scholar] [CrossRef]
- Celic, I.; Verreault, A.; Boeke, J.D. Histone H3 K56 hyperacetylation perturbs replisomes and causes DNA damage. Genetics 2008, 179, 1769–1784. [Google Scholar] [CrossRef]
- Fillingham, J.; Greenblatt, J.F. A histone code for chromatin assembly. Cell 2008, 134, 206–208. [Google Scholar] [CrossRef]
- Andersen, M.P.; Nelson, Z.W.; Hetrick, E.D.; Gottschling, D.E. A genetic screen for increased loss of heterozygosity in Saccharomyces cerevisiae. Genetics 2008, 179, 1179–1195. [Google Scholar] [CrossRef] [PubMed]
- Kadyrova, L.Y.; Mertz, T.M.; Zhang, Y.; Northam, M.R.; Sheng, Z.; Lobachev, K.S.; Shcherbakova, P.V.; Kadyrov, F.A. A reversible histone H3 acetylation cooperates with mismatch repair and replicative polymerases in maintaining genome stability. PLoS Genet. 2013, 9, e1003899. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Miller, A.; Kirchmaier, A.L. HST3/HST4-dependent deacetylation of lysine 56 of histone H3 in silent chromatin. Mol. Biol. Cell 2008, 19, 4993–5005. [Google Scholar] [CrossRef][Green Version]
- Munoz-Galvan, S.; Jimeno, S.; Rothstein, R.; Aguilera, A. Histone H3K56 acetylation, Rad52, and non-DNA repair factors control double-strand break repair choice with the sister chromatid. PLoS Genet. 2013, 9, e1003237. [Google Scholar] [CrossRef]
- Gershon, L.; Kupiec, M. A novel role for Dun1 in the regulation of origin firing upon hyper-acetylation of H3K56. PLoS Genet. 2021, 17, e1009391. [Google Scholar] [CrossRef]
- Dershowitz, A.; Snyder, M.; Sbia, M.; Skurnick, J.H.; Ong, L.Y.; Newlon, C.S. Linear derivatives of Saccharomyces cerevisiae chromosome III can be maintained in the absence of autonomously replicating sequence elements. Mol. Cell. Biol. 2007, 27, 4652–4663. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Myung, K.; Donaldson, A.D. Is PCNA unloading the central function of the Elg1/ATAD5 replication factor C-like complex? Cell Cycle 2013, 12, 2570–2579. [Google Scholar] [CrossRef]
- Shemesh, K.; Sebesta, M.; Pacesa, M.; Sau, S.; Bronstein, A.; Parnas, O.; Liefshitz, B.; Venclovas, C.; Krejci, L.; Kupiec, M. A structure-function analysis of the yeast Elg1 protein reveals the importance of PCNA unloading in genome stability maintenance. Nucleic Acids Res. 2017, 45, 3189–3203. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.Z.; Schnakenberg, S.L.; Zakian, V.A. Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: Viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol. Cell. Biol. 2004, 24, 3198–3212. [Google Scholar] [CrossRef]
- Luciano, P.; Dehe, P.M.; Audebert, S.; Geli, V.; Corda, Y. Replisome function during replicative stress is modulated by histone h3 lysine 56 acetylation through Ctf4. Genetics 2015, 199, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; Jones, R.C.; Sanchez-Diaz, A.; Kanemaki, M.; van Deursen, F.; Edmondson, R.D.; Labib, K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006, 8, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; van Deursen, F.; Polychronopoulos, D.; Foltman, M.; Jones, R.C.; Edmondson, R.D.; Calzada, A.; Labib, K. A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J. 2009, 28, 2992–3004. [Google Scholar] [CrossRef]
- Lengronne, A.; Pasero, P. Closing the MCM cycle at replication termination sites. EMBO Rep. 2014, 15, 1226–1227. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moriel-Carretero, M.; Pasero, P.; Pardo, B. DDR Inc., one business, two associates. Curr Genet 2019, 65, 445–451. [Google Scholar] [CrossRef]
- Gispan, A.; Carmi, M.; Barkai, N. Checkpoint-independent scaling of the Saccharomyces cerevisiae DNA replication program. BMC Biol. 2014, 12, 79. [Google Scholar] [CrossRef]
- Poli, J.; Tsaponina, O.; Crabbe, L.; Keszthelyi, A.; Pantesco, V.; Chabes, A.; Lengronne, A.; Pasero, P. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 2012, 31, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Diffley, J.F. MCM: One ring to rule them all. Curr. Opin. Struct. Biol. 2016, 37, 145–151. [Google Scholar] [CrossRef]
- Green, C.M.; Almouzni, G. When repair meets chromatin. First in series on chromatin dynamics. EMBO Rep. 2002, 3, 28–33. [Google Scholar] [CrossRef]
- Seeber, A.; Dion, V.; Gasser, S.M. Checkpoint kinases and the INO80 nucleosome remodeling complex enhance global chromatin mobility in response to DNA damage. Genes Dev. 2013, 27, 1999–2008. [Google Scholar] [CrossRef]
- Weinert, T.A.; Hartwell, L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 1988, 241, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Crabbe, L.; Pasero, P. Signaling pathways of replication stress in yeast. FEMS Yeast Res. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Sau, S.; Kupiec, M. A role for the yeast PCNA unloader Elg1 in eliciting the DNA damage checkpoint. Curr. Genet. 2020, 66, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Sau, S.; Liefshitz, B.; Kupiec, M. The Yeast PCNA Unloader Elg1 RFC-Like Complex Plays a Role in Eliciting the DNA Damage Checkpoint. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A.; Lowndes, N.F.; Jackson, S.P. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000, 408, 1001–1004. [Google Scholar] [CrossRef]
- Ciardo, D.; Goldar, A.; Marheineke, K. On the Interplay of the DNA Replication Program and the Intra-S Phase Checkpoint Pathway. Genes 2019, 10, 94. [Google Scholar] [CrossRef]
- Clemenson, C.; Marsolier-Kergoat, M.C. DNA damage checkpoint inactivation: Adaptation and recovery. DNA Repair 2009, 8, 1101–1109. [Google Scholar] [CrossRef]
- Hastie, C.J.; Vazquez-Martin, C.; Philp, A.; Stark, M.J.; Cohen, P.T. The Saccharomyces cerevisiae orthologue of the human protein phosphatase 4 core regulatory subunit R2 confers resistance to the anticancer drug cisplatin. FEBS J. 2006, 273, 3322–3334. [Google Scholar] [CrossRef]
- Simoneau, A.; Delgoshaie, N.; Celic, I.; Dai, J.; Abshiru, N.; Costantino, S.; Thibault, P.; Boeke, J.D.; Verreault, A.; Wurtele, H. Interplay between histone H3 lysine 56 deacetylation and chromatin modifiers in response to DNA damage. Genetics 2015, 200, 185–205. [Google Scholar] [CrossRef]
- Tsabar, M.; Waterman, D.P.; Aguilar, F.; Katsnelson, L.; Eapen, V.V.; Memisoglu, G.; Haber, J.E. Asf1 facilitates dephosphorylation of Rad53 after DNA double-strand break repair. Genes Dev. 2016, 30, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Carson, J.J.; Feser, J.; Tamburini, B.; Zabaronick, S.; Linger, J.; Tyler, J.K. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 2008, 134, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Hombauer, H.; Srivatsan, A.; Putnam, C.D.; Kolodner, R.D. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 2011, 334, 1713–1716. [Google Scholar] [CrossRef]
- Rodriges Blanko, E.; Kadyrova, L.Y.; Kadyrov, F.A. DNA Mismatch Repair Interacts with CAF-1- and ASF1A-H3-H4-dependent Histone (H3-H4)2 Tetramer Deposition. J. Biol. Chem. 2016, 291, 9203–9217. [Google Scholar] [CrossRef]
- Li, F.; Tian, L.; Gu, L.; Li, G.M. Evidence that nucleosomes inhibit mismatch repair in eukaryotic cells. J. Biol. Chem. 2009, 284, 33056–33061. [Google Scholar] [CrossRef]
- Che, J.; Smith, S.; Kim, Y.J.; Shim, E.Y.; Myung, K.; Lee, S.E. Hyper-Acetylation of Histone H3K56 Limits Break-Induced Replication by Inhibiting Extensive Repair Synthesis. PLoS Genet. 2015, 11, e1004990. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, N.; Goldfarb, T.; Studamire, B.; Alani, E.; Haber, J.E. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. USA 2004, 101, 9315–9320. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, U.; Alani, E. Understanding how mismatch repair proteins participate in the repair/anti-recombination decision. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, U.; Mackenroth, B.; Shalloway, D.; Alani, E. Chromatin Modifiers Alter Recombination Between Divergent DNA Sequences. Genetics 2019, 212, 1147–1162. [Google Scholar] [CrossRef] [PubMed]
- Newman, T.J.; Mamun, M.A.; Nieduszynski, C.A.; Blow, J.J. Replisome stall events have shaped the distribution of replication origins in the genomes of yeasts. Nucleic Acids Res. 2013, 41, 9705–9718. [Google Scholar] [CrossRef]
- Feng, B.; Lin, Y.; Zhou, L.; Guo, Y.; Friedman, R.; Xia, R.; Hu, F.; Liu, C.; Tang, J. Reconstructing Yeasts Phylogenies and Ancestors from Whole Genome Data. Sci. Rep. 2017, 7, 15209. [Google Scholar] [CrossRef]
- Theis, J.F.; Irene, C.; Dershowitz, A.; Brost, R.L.; Tobin, M.L.; di Sanzo, F.M.; Wang, J.Y.; Boone, C.; Newlon, C.S. The DNA damage response pathway contributes to the stability of chromosome III derivatives lacking efficient replicators. PLoS Genet. 2010, 6, e1001227. [Google Scholar] [CrossRef]
- Kovtun, I.V.; McMurray, C.T. Features of trinucleotide repeat instability in vivo. Cell Res. 2008, 18, 198–213. [Google Scholar] [CrossRef]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef]
- Fouche, N.; Ozgur, S.; Roy, D.; Griffith, J.D. Replication fork regression in repetitive DNAs. Nucleic Acids Res. 2006, 34, 6044–6050. [Google Scholar] [CrossRef][Green Version]
- Pelletier, R.; Krasilnikova, M.M.; Samadashwily, G.M.; Lahue, R.; Mirkin, S.M. Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 2003, 23, 1349–1357. [Google Scholar] [CrossRef]
- McMurray, C.T. Hijacking of the mismatch repair system to cause CAG expansion and cell death in neurodegenerative disease. DNA Repair 2008, 7, 1121–1134. [Google Scholar] [CrossRef]
- Clemente-Ruiz, M.; Gonzalez-Prieto, R.; Prado, F. Histone H3K56 acetylation, CAF1, and Rtt106 coordinate nucleosome assembly and stability of advancing replication forks. PLoS Genet. 2011, 7, e1002376. [Google Scholar] [CrossRef]
- Yang, J.H.; Freudenreich, C.H. The Rtt109 histone acetyltransferase facilitates error-free replication to prevent CAG/CTG repeat contractions. DNA Repair 2010, 9, 414–420. [Google Scholar] [CrossRef]
- Freudenreich, C.H.; Stavenhagen, J.B.; Zakian, V.A. Stability of a CTG/CAG trinucleotide repeat in yeast is dependent on its orientation in the genome. Mol. Cell. Biol. 1997, 17, 2090–2098. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, J.; Li, Q. The replisome guides nucleosome assembly during DNA replication. Cell Biosci. 2020, 10, 37. [Google Scholar] [CrossRef]
- Prado, F.; Clemente-Ruiz, M. Nucleosome assembly and genome integrity: The fork is the link. Bioarchitecture 2012, 2, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Lucchini, R.; Wellinger, R.E.; Sogo, J.M. Nucleosome positioning at the replication fork. EMBO J. 2001, 20, 7294–7302. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef]
- Kramara, J.; Osia, B.; Malkova, A. Break-Induced Replication: The Where, The Why, and The How. Trends Genet. TIG 2018, 34, 518–531. [Google Scholar] [CrossRef]
- Malkova, A.; Naylor, M.L.; Yamaguchi, M.; Ira, G.; Haber, J.E. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol. Cell. Biol. 2005, 25, 933–944. [Google Scholar] [CrossRef]
- Signon, L.; Malkova, A.; Naylor, M.L.; Klein, H.; Haber, J.E. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol. Cell. Biol. 2001, 21, 2048–2056. [Google Scholar] [CrossRef]
- Feldman, J.L.; Peterson, C.L. Yeast Sirtuin Family Members Maintain Transcription Homeostasis to Ensure Genome Stability. Cell Rep. 2019, 27, 2978–2989.e5. [Google Scholar] [CrossRef]
- Jung, I.; Seo, J.; Lee, H.S.; Stanton, L.W.; Kim, D.; Choi, J.K. Global mapping of the regulatory interactions of histone residues. FEBS Lett. 2015, 589, 4061–4070. [Google Scholar] [CrossRef]
- Minard, L.V.; Williams, J.S.; Walker, A.C.; Schultz, M.C. Transcriptional regulation by Asf1: New mechanistic insights from studies of the DNA damage response to replication stress. J. Biol. Chem. 2011, 286, 7082–7092. [Google Scholar] [CrossRef]
- Voichek, Y.; Bar-Ziv, R.; Barkai, N. A role for Rtt109 in buffering gene-dosage imbalance during DNA replication. Nucleus 2016, 7, 375–381. [Google Scholar] [CrossRef][Green Version]
- Dodson, A.E.; Rine, J. Heritable capture of heterochromatin dynamics in Saccharomyces cerevisiae. eLife 2015, 4, e05007. [Google Scholar] [CrossRef] [PubMed]
- Oppikofer, M.; Kueng, S.; Martino, F.; Soeroes, S.; Hancock, S.M.; Chin, J.W.; Fischle, W.; Gasser, S.M. A dual role of H4K16 acetylation in the establishment of yeast silent chromatin. EMBO J. 2011, 30, 2610–2621. [Google Scholar] [CrossRef]
- Han, J.; Zhou, H.; Li, Z.; Xu, R.M.; Zhang, Z. Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J. Biol. Chem. 2007, 282, 28587–28596. [Google Scholar] [CrossRef] [PubMed]
- Ide, S.; Saka, K.; Kobayashi, T. Rtt109 prevents hyper-amplification of ribosomal RNA genes through histone modification in budding yeast. PLoS Genet. 2013, 9, e1003410. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, S.; Botsios, S.; Donaldson, A.D. Histone H3 lysine 56 acetylation by Rtt109 is crucial for chromosome positioning. J. Cell Biol. 2008, 183, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Hachinohe, M.; Hanaoka, F.; Masumoto, H. Hst3 and Hst4 histone deacetylases regulate replicative lifespan by preventing genome instability in Saccharomyces cerevisiae. Genes Cells Devoted Mol. Cell. Mech. 2011, 16, 467–477. [Google Scholar] [CrossRef]
- Lamming, D.W.; Latorre-Esteves, M.; Medvedik, O.; Wong, S.N.; Tsang, F.A.; Wang, C.; Lin, S.J.; Sinclair, D.A. HST2 mediates SIR2-independent life-span extension by calorie restriction. Science 2005, 309, 1861–1864. [Google Scholar] [CrossRef]
- Saredi, G.; Huang, H.; Hammond, C.M.; Alabert, C.; Bekker-Jensen, S.; Forne, I.; Reveron-Gomez, N.; Foster, B.M.; Mlejnkova, L.; Bartke, T.; et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 2016, 534, 714–718. [Google Scholar] [CrossRef]
- Lo, K.A.; Bauchmann, M.K.; Baumann, A.P.; Donahue, C.J.; Thiede, M.A.; Hayes, L.S.; des Etages, S.A.; Fraenkel, E. Genome-wide profiling of H3K56 acetylation and transcription factor binding sites in human adipocytes. PLoS ONE 2011, 6, e19778. [Google Scholar] [CrossRef] [PubMed]
- Aves, S.J.; Liu, Y.; Richards, T.A. Evolutionary diversification of eukaryotic DNA replication machinery. Sub-Cell. Biochem. 2012, 62, 19–35. [Google Scholar] [CrossRef]
- Yuan, H.; Su, L.; Chen, W.Y. The emerging and diverse roles of sirtuins in cancer: A clinical perspective. OncoTargets Ther. 2013, 6, 1399–1416. [Google Scholar] [CrossRef]
- Mirabella, A.C.; Foster, B.M.; Bartke, T. Chromatin deregulation in disease. Chromosoma 2016, 125, 75–93. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Category | Phenotype | Source |
|---|---|---|
| Sensitivity to genotoxic agents: | Campthotecin (CPT) | [13] |
| Bleomycin (ionizing radiation) | ||
| Ultraviolet radiation (UV) | [40] | |
| Methyl Methanesulfonate (MMS) | [41] | |
| Hydroxyurea (HU) | ||
| Peroxide (H2O2) | [51] | |
| Genotoxic stress markers: | Rad52-foci accumulation | [48] |
| Ddc2-foci accumulation | ||
| Spontaneous Rad53 phosphorylation | [52] | |
| Genome stability: | Increased loss of heterozygosity | [54] |
| Elevated recombination rates | [55] | |
| Gross chromosomal rearrangements | ||
| Small insertions/deletions | ||
| Elevated rates of nondisjunction events | ||
| Chromatin changes: | Derepression of subtelomeric genes | [56] |
| Sister chromatid cohesion defects | [48] | |
| Other: | G2/M cycle arrest/delay | [40] |
| Thermosensitivity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gershon, L.; Kupiec, M. The Amazing Acrobat: Yeast’s Histone H3K56 Juggles Several Important Roles While Maintaining Perfect Balance. Genes 2021, 12, 342. https://doi.org/10.3390/genes12030342
Gershon L, Kupiec M. The Amazing Acrobat: Yeast’s Histone H3K56 Juggles Several Important Roles While Maintaining Perfect Balance. Genes. 2021; 12(3):342. https://doi.org/10.3390/genes12030342
Chicago/Turabian StyleGershon, Lihi, and Martin Kupiec. 2021. "The Amazing Acrobat: Yeast’s Histone H3K56 Juggles Several Important Roles While Maintaining Perfect Balance" Genes 12, no. 3: 342. https://doi.org/10.3390/genes12030342
APA StyleGershon, L., & Kupiec, M. (2021). The Amazing Acrobat: Yeast’s Histone H3K56 Juggles Several Important Roles While Maintaining Perfect Balance. Genes, 12(3), 342. https://doi.org/10.3390/genes12030342