
Application of Genomics to Understand Salt Tolerance in Lentil




Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotyping
2.2. Probe Designing for Lentil Targeted-GBS (tGBS) Method
2.3. tGBS Library Preparation, Sequencing and Variant Calling
2.4. Transcriptome-based GBS Library Preparation, Sequencing and Variant Calling
2.5. GWAS, Candidate Genes Identification and Pedigree Haplotype Analysis
2.6. Understanding Salt Tolerance Mechanism in Lentil Using Elemental Analysis
3. Results
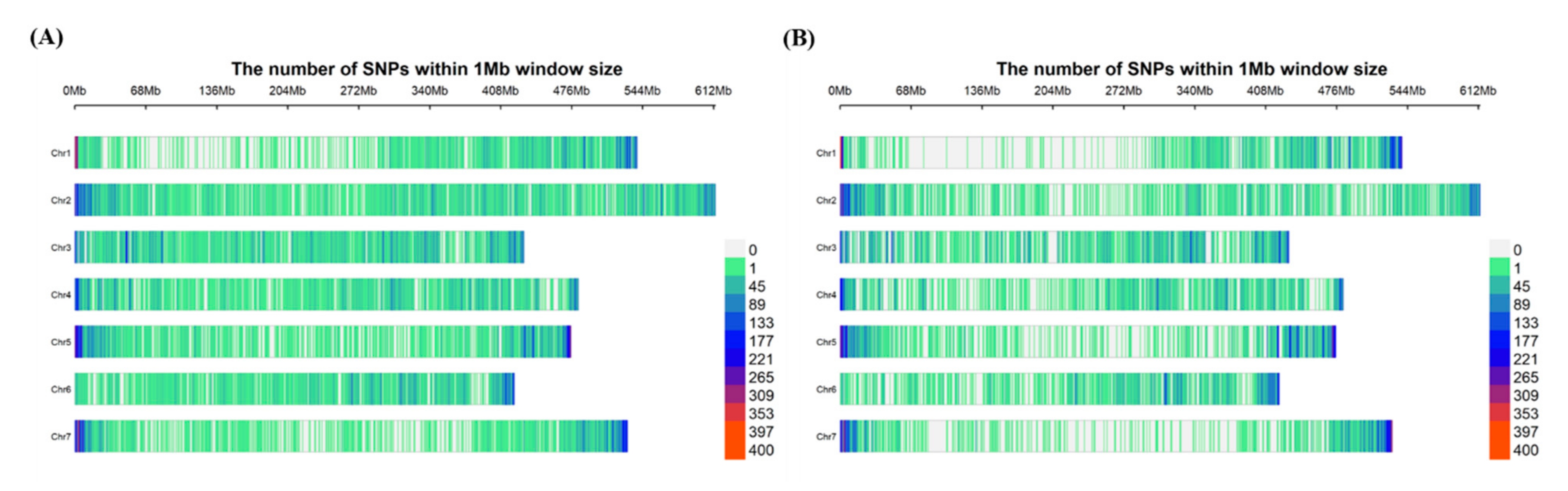
3.1. Evaluation of SNP Markers Captured in Novel tGBS Method and GBS-t Method
3.2. Model Selection for Marker-Trait Association Study
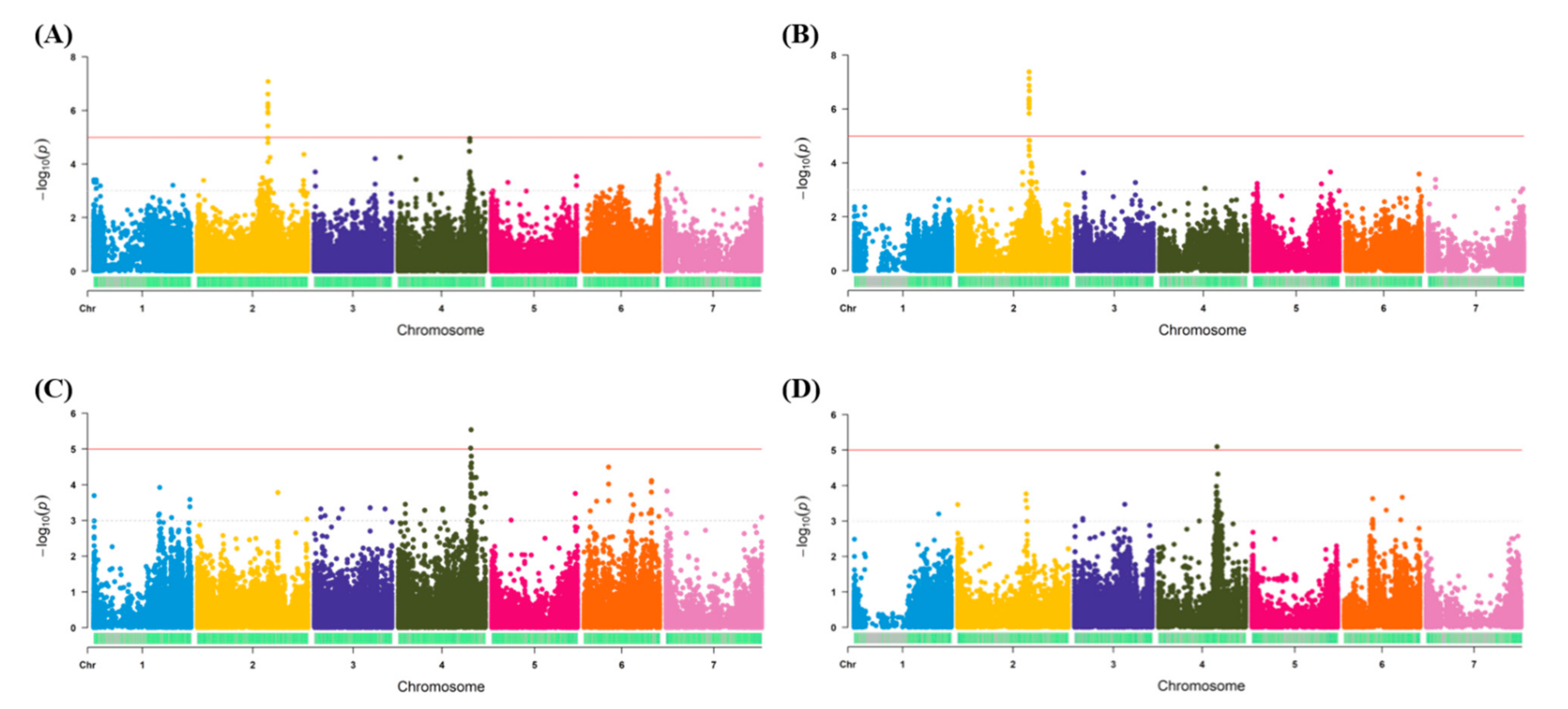
3.3. Regions Identified for Salt Tolerance Traits Using Different GBS Methods
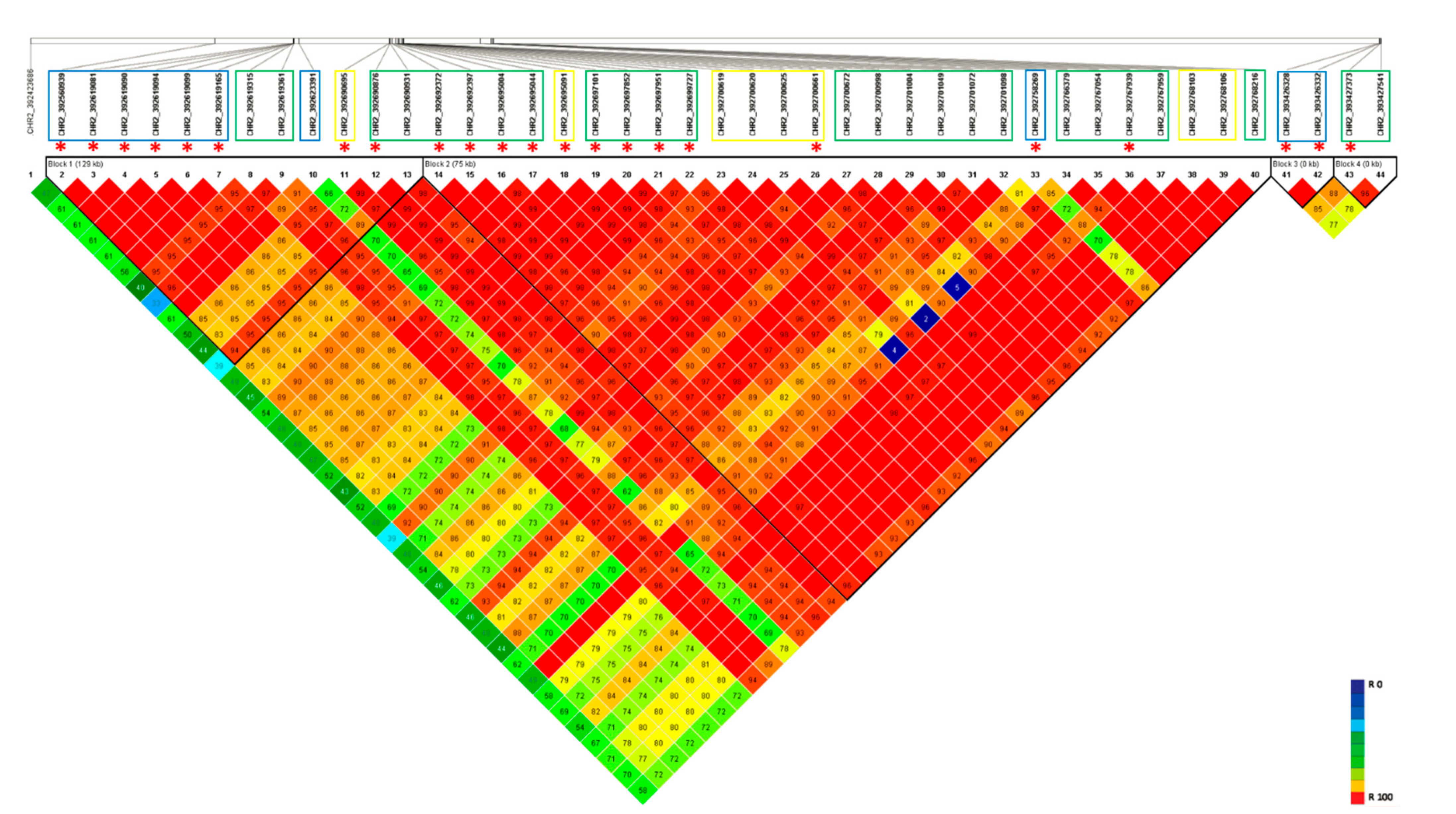
3.4. Haplotype Blocks on Chromosome 2
3.5. Candidate Genes Identified for Genomic Regions
3.6. Haplotype Variation on Chromosome 2
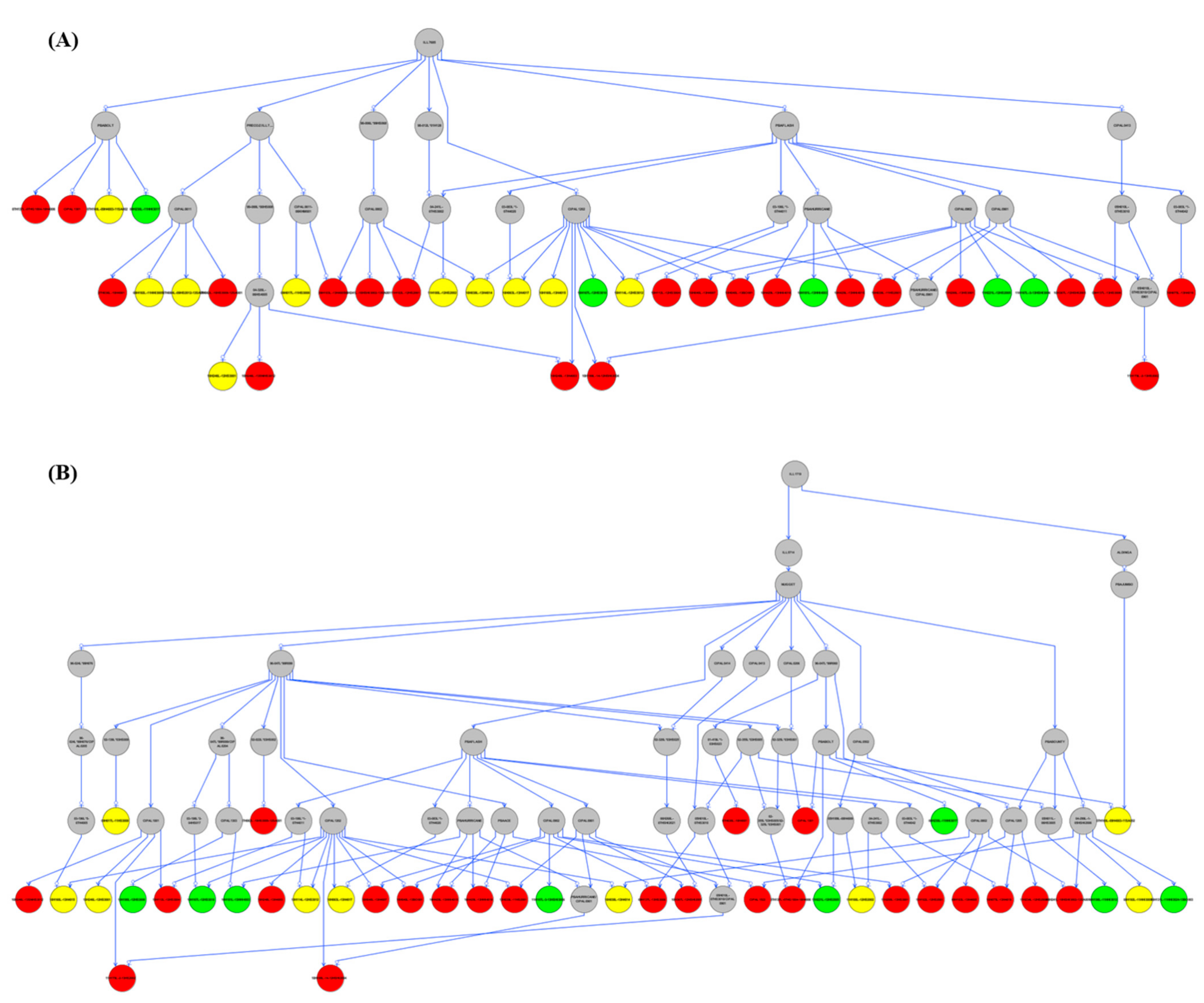
3.7. Pedigree Analysis
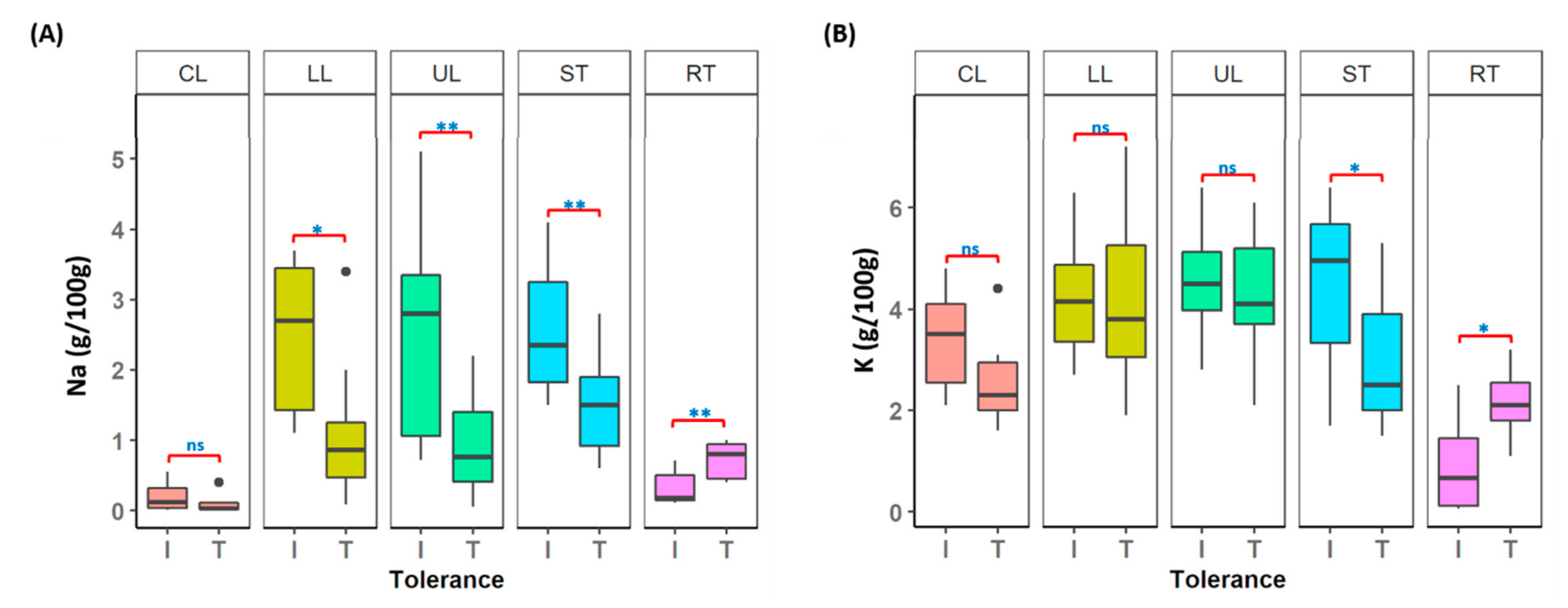
3.8. Understanding Salt Tolerance Mechanism in Lentil
4. Discussion
4.1. Identification of Genomic Regions Conferring Salt Tolerance in Lentil
4.2. Breeding for Salt Tolerance, Haplotypes, and Pedigree Analysis
4.3. Potential Candidate Genes and Salt Tolerance Mechanism in Lentil
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soren, K.R.; Madugula, P.; Kumar, N.; Barmukh, R.; Sengar, M.S.; Bharadwaj, C.; Sharma, P.C.; Singh, S.; Bhandari, A.; Singh, J.; et al. Genetic dissection and identification of candidate genes for salinity tolerance using Axiom®CicerSNP array in chickpea. Int. J. Mol. Sci. 2020, 21, 5058. [Google Scholar] [CrossRef]
- Kumawat, K.R.; Gothwal, D.K.; Singh, D. Salinity tolerance of lentil genotypes based on stress tolerance indices. J. Pharmacogn. Phytochem. 2017, 6, 1368–1372. [Google Scholar]
- Spies, B.; Woodgate, P. Salinity and Hydrogeology. In Salinity Mapping Methods in the Australian Context; Department of the Environment and Heritage and Agriculture, Fisheries and Forestry: Canberra, Australia, 2005; p. 17. [Google Scholar]
- Maher, L.; Armstrong, R.; Connor, D. Salt Tolerant Lentils-A Possibility for the Future. In Solutions for a Better Environment, Proceedings of the 11th Australian Agronomy Conference, Geelong, VIC, Australia, 2–6 February 2003; Unkovich, M., O’Leary, G., Eds.; The Australian Society of Agronomy Inc.: Geelong, Australia, 2003; Available online: http://www.agronomyaustraliaproceedings.org/images/sampledata/2003/c/17/maher.pdf (accessed on 21 October 2019).
- FAOSTAT. Available online: http://www.fao.org/faostat/en/#data (accessed on 11 February 2021).
- Tesfaye, A.; Petros, Y.; Zeleke, H. Screening some accessions of lentil (Lens culinaris M.) for salt tolerance at germination and early seedling stage in Eastern Ethiopia. Int. J. Technol. Enhanc. Emerg. Eng. Res. 2014, 2, 106–113. [Google Scholar]
- Jayasundara, H.P.S.; Thomson, B.D.; Tang, C. Responses of cool season grain legumes to soil abiotic stresses. Adv. Agron. 1998, 63, 77–151. [Google Scholar]
- Kökten, K.; Karaköy, T.; Bakoğlu, A.; Akçura, M. Determination of salinity tolerance of some lentil (Lens culinaris M.) varieties. J. Food Agric. Environ. 2010, 8, 140–143. [Google Scholar]
- Roy, S.J.; Negrao, S.; Tester, M. Salt resistant crop plants. Curr. Opin. Biotechnol. 2014, 26, 115–124. [Google Scholar] [CrossRef]
- Nadeem, M.; Li, J.; Yahya, M.; Wang, M.; Ali, A.; Cheng, A.; Wang, X.; Ma, C. Grain legumes and fear of salt stress: Focus on mechanisms and management strategies. Int. J. Mol. Sci. 2019, 20, 799. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, P.M.; Bressan, R.A. Plant cellular and molecular responses to high salinity. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 2000, 51, 463–499. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Hussain, M.; Wakeel, A.; Siddique, K.H.M. Salt stress in maize: Effects, resistance mechanisms, and management. A review. Agron. Sustain. Dev. 2015, 35, 461–481. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant. Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Qurashi, A.W.; Sabri, A.N. Osmolyte accumulation in moderately halophilic bacteria improves salt tolerance of chickpea. Pak. J. Bot. 2013, 45, 1011–1016. [Google Scholar]
- El Sabagh, A.; Sorour, S.; Omar, A.E.; Ragab, A.; Islam, M.S.; Ueda, A.; Saneoka, H. Alleviation of adverse effects of salt stress on soybean (Glycine max. L.) by using osmoprotectants and organic nutrients. Int. J. Innov. Res. Sci. Eng. 2015, 9, 921–925. [Google Scholar]
- Vishwakarma, K.; Upadhyay, N.; Kumar, N.; Yadav, G.; Singh, J.; Mishra, R.K.; Kumar, V.; Verma, R.; Upadhyay, R.G.; Pandey, M.; et al. Abscisic acid signaling and abiotic stress tolerance in plants: A review on current knowledge and future prospects. Front. Plant. Sci. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Turan, M.A.; Turkmen, N.; Taban, N. Effect of NaCl on stomatal resistance and proline, chlorophyll, Na, Cl, and K concentration of lentil plants. J. Agron. 2007, 6, 378–381. [Google Scholar] [CrossRef]
- Singh, D.; Singh, C.K.; Kumari, S.; Tomar, R.S.S.; Karwa, S.; Singh, R.; Singh, R.B.; Sarkar, S.K.; Pal, M. Discerning morpho-anatomical, physiological and molecular multiformity in cultivated and wild genotypes of lentil with reconciliation to salinity stress. PLoS ONE 2017, 12, e0177465. [Google Scholar] [CrossRef]
- Pandey, A.K.; Sengar, R.S. Effect of salt stress on salt tolerant indices of morpho-physiological traits and yield attributes of lentil (Lens culinaris Medik.). Int. J. Chem. Stud. 2020, 8, 2292–2301. [Google Scholar] [CrossRef][Green Version]
- Medina, C.A.; Hawkins, C.; Liu, X.-P.; Peel, M.; Yu, L.-X. Genome-wide association and prediction of traits related to salt tolerance in autotetraploid alfalfa (Medicago sativa L.). Int. J. Mol. Sci. 2020, 21, 3361. [Google Scholar] [CrossRef]
- Tafesse, E.G.; Gali, K.K.; Lachagari, V.B.R.; Bueckert, R.; Warkentin, T.D. Genome-wide association mapping for heat stress responsive traits in field pea. Int. J. Mol. Sci. 2020, 21, 2043. [Google Scholar] [CrossRef] [PubMed]
- Patishtan, J.; Hartley, T.N.; Fonseca de Carvalho, R.; Maathuis, F.J.M. Genome-wide association studies to identify rice salt-tolerance markers. Plant. Cell Environ. 2018, 41, 970–982. [Google Scholar] [CrossRef]
- Li, N.; Zheng, H.; Cui, J.; Wang, J.; Liu, H.; Sun, J.; Liu, T.; Zhao, H.; Lai, Y.; Zou, D. Genome-wide association study and candidate gene analysis of alkalinity tolerance in japonica rice germplasm at the seedling stage. Rice 2019, 12, 24. [Google Scholar] [CrossRef]
- Lekklar, C.; Pongpanich, M.; Suriya-Arunroj, D.; Chinpongpanich, A.; Tsai, H.; Comai, L.; Chadchawan, S.; Buaboocha, T. Genome-wide association study for salinity tolerance at the flowering stage in a panel of rice accessions from Thailand. BMC Genom. 2019, 20, 76. [Google Scholar] [CrossRef]
- Yuan, J.; Wang, X.; Zhao, Y.; Khan, N.U.; Zhao, Z.; Zhang, Y.; Wen, X.; Tang, F.; Wang, F.; Li, Z. Genetic basis and identification of candidate genes for salt tolerance in rice by GWAS. Sci. Rep. 2020, 10, 9958. [Google Scholar] [CrossRef]
- Sun, Z.; Li, H.; Zhang, Y.; Li, Z.; Ke, H.; Wu, L.; Zhang, G.; Wang, X.; Ma, Z. Identification of SNPs and candidate genes associated with salt tolerance at the seedling stage in cotton (Gossypium hirsutum L.). Front. Plant. Sci. 2018, 9, 1011. [Google Scholar] [CrossRef]
- Dilnur, T.; Peng, Z.; Pan, Z.; Palanga, K.K.; Jia, Y.; Gong, W.; Du, X. Association analysis of salt tolerance in Asiatic cotton (Gossypium arboretum) with SNP markers. Int. J. Mol. Sci. 2019, 20, 2168. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Chen, P.; Korth, K.; Hancock, F.; Pereira, A.; Brye, K.; Wu, C.; Shi, A. Genome-wide association study (GWAS) of salt tolerance in worldwide soybean germplasm lines. Mol. Breed. 2017, 37, 30. [Google Scholar] [CrossRef]
- Luo, X.; Wang, B.; Gao, S.; Zhang, F.; Terzaghi, W.; Dai, M. Genome-wide association study dissects the genetic bases of salt tolerance in maize seedlings. J. Integr. Plant. Biol. 2019, 61, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, M.M.; Pembleton, L.P.; Baillie, R.C.; Drayton, M.C.; Sudheesh, S.; Kaur, S.; Shinozuka, H.; Verma, P.; Spangenberg, G.C.; Daetwyler, H.D.; et al. Genotyping-by-sequencing through transcriptomics: Implementation in a range of crop species with varying reproductive habits and ploidy levels. Plant. Biotechnol. J. 2018, 16, 877–889. [Google Scholar] [CrossRef]
- Lesur, I.; Alexandre, H.; Boury, C.; Chancerel, E.; Plomion, C.; Kremer, A. Development of target sequence capture and estimation of genomic relatedness in a mixed oak stand. Front. Plant. Sci. 2018, 9, 996. [Google Scholar] [CrossRef]
- Hale, H.; Gardner, E.M.; Viruel, J.; Pokorny, L.; Johnson, M.G. Strategies for reducing per-sample costs in target capture sequencing for phylogenomics and population genomics in plants. Appl. Plant Sci. 2020, 8, e11337. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Guo, H.; Kong, W.; Chandnani, R.; Shuang, L.; Paterson, A.H. Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant. Sci. 2016, 242, 14–22. [Google Scholar] [CrossRef]
- Dashti, M.J.S.; Gamieldien, J. A practical guide to filtering and prioritizing genetic variants. Biotechniques 2017, 62, 18–30. [Google Scholar] [CrossRef]
- Andermann, T.; Torres Jiménez, M.F.; Matos-Maraví, P.; Batista, R.; Blanco-Pastor, J.L.; Gustafsson, A.L.S.; Kistler, L.; Liberal, I.M.; Oxelman, B.; Bacon, C.D.; et al. A guide to carrying out a phylogenomic target sequence capture project. Front. Genet. 2020, 10, 1407. [Google Scholar] [CrossRef]
- Wong, M.M.L.; Gujaria-Verma, N.; Ramsay, L.; Yuan, H.Y.; Caron, C.; Diapari, M.; Vandenberg, A.; Bett, K.E. Classification and characterization of species within the genus lens using genotyping-by-sequencing (GBS). PLoS ONE 2015, 10, e0122025. [Google Scholar] [CrossRef]
- Ogutcen, E.; Ramsay, L.; von Wettberg, E.B.; Bett, K.E. Capturing variation in Lens (Fabaceae): Development and utility of an exome capture array for lentil. Appl. Plant. Sci. 2018, 6, e01165. [Google Scholar] [CrossRef]
- Dissanayake, R.; Braich, S.; Cogan, N.O.I.; Smith, K.; Kaur, S. Characterization of genetic and allelic diversity amongst culti-vated and wild lentil accessions for germplasm enhancement. Front. Genet. 2020, 11, 546. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, L.; Koh, C.; Konkin, D.; Cook, D.; Penmetsa, V.; Dongying, G.; Coyne, C.; Humann, J.; Kaur, S.; Dolezel, J.; et al. Lens culinaris CDC Redberry Genome Assembly v2.0. 2019. Available online: https://knowpulse.usask.ca/genome-assembly/Lcu.2RBY (accessed on 10 September 2019).
- Dissanayake, R.; Kahrood, H.V.; Dimech, A.M.; Noy, D.M.; Rosewarne, G.M.; Smith, K.F.; Cogan, N.O.I.; Kaur, S. Development and application of image-based high-throughput phenotyping methodology for salt tolerance in lentils. Agronomy 2020, 10, 1992. [Google Scholar] [CrossRef]
- Dissanayake, R.; Pembleton, L.; Cogan, N.O.I.; Smith, K.F.; Kaur, S. Novel Targeted Genotyping by Sequencing (tGBS): A Promising Tool for Lentil Genetics Research and Breeding. Available online: https://apc2019.com.au/abstracts/pdf/abstract_9.pdf (accessed on 25 October 2019).
- Sudheesh, S.; Verma, P.; Forster, J.W.; Cogan, N.O.I.; Kaur, S. Generation and characterisation of a reference transcriptome for lentil (Lens culinaris Medik.). Int. J. Mol. Sci. 2016, 17, 1887. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, L.; Chan, C.; Sharpe, A.G.; Cook, D.R.; Penmetsa, R.V.; Chang, P.; Coyne, C.; McGee, R.; Main, D.; Edwards, D.; et al. Lens culinaris CDC Redberry Genome Assembly v1.2. 2016. Available online: https://knowpulse.usask.ca/genome-assembly/Lc1.2. (accessed on 15 November 2018).
- NuGEN. Available online: https://www.nugen.com/ (accessed on 15 November 2018).
- TrimGalore. Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 16 June 2019).
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Pembleton, L.W.; Cogan, N.O.I.; Forster, J.W. StAMPP: An R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 2013, 13, 946–952. [Google Scholar] [CrossRef]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software. 2006. Available online: http://darwin.cirad.fr/ (accessed on 10 July 2019).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 13 July 2019).
- Yin, L. CMplot. 2020. Available online: https://github.com/YinLiLin/CMplot (accessed on 6 February 2020).
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef]
- Pulse Breeding Australia. Available online: https://www.pbseeds.com.au/docs/ (accessed on 13 August 2020).
- Shaw, P.D.; Graham, M.; Kennedy, J.; Milne, I.; Marshall, D.F. Helium: Visualization of large scale plant pedigrees. BMC Bioinform. 2014, 15, 259. [Google Scholar] [CrossRef]
- Kalra, Y.P. Handbook of Reference Methods for Plant. Analysis; CRC Press: Boca Raton, FL, USA, 1998; pp. 37–170. [Google Scholar]
- Kassambara, A. ggpubr. 2020. Available online: https://rdrr.io/cran/ggpubr/ (accessed on 10 September 2020).
- Morton, M.J.L.; Awlia, M.; Al-Tamimi, N.; Saade, S.; Pailles, Y.; Negrào, S.; Tester, M. Salt stress under the scalpel-Dissecting the genetics of salt tolerance. Plant. J. 2019, 97, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Singh, C.K.; Tomar, R.S.S.; Sharma, S.; Karwa, S.; Pal, M.; Singh, V.; Sanwal, S.K.; Sharma, P.C. Genetics and molecular mapping for salinity stress tolerance at seedling stage in lentil (Lens culinaris Medik). Crop. Sci. 2020, 60, 1254–1266. [Google Scholar] [CrossRef]
- Malmberg, M.M.; Shi, F.; Spangenberg, G.C.; Daetwyler, H.D.; Cogan, N. Diversity and genome analysis of australian and global oilseed Brassica napus L. germplasm using transcriptomics and whole genome re-sequencing. Front. Plant. Sci. 2018, 9, 508. [Google Scholar] [CrossRef]
- Rahman, M.A.; Bimpong, I.K.; Bizimana, J.B.; Pascual, E.D.; Arceta, M.; Swamy, B.; Diaw, F.; Rahman, M.S.; Singh, R.K. Mapping QTLs using a novel source of salinity tolerance from Hasawi and their interaction with environments in rice. Rice 2017, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Zheng, Q.; Luo, Q.; Teng, W.; Li, H.; Li, B.; Li, Z. Genome-wide association study of yield and related traits in common wheat under salt-stress conditions. BMC Plant. Biol. 2021, 21, 27. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop. J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- MacLeod, I.M.; Bowman, P.J.; Vander Jagt, C.J.; Haile-Mariam, M.; Kemper, K.E.; Chamberlain, A.J.; Schrooten, C.; Hayes, B.J.; Goddard, M.E. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Fikere, M.; Barbulescu, D.M.; Malmberg, M.M.; Shi, F.; Joshua, J.C.O.; Slater, A.T.; MacLeod, I.M.; Bowman, P.J.; Salisbury, P.A.; Spangenberg, G.C.; et al. Genomic prediction using prior quantitative trait loci information reveals a large reservoir of underutilised blackleg resistance in diverse canola (Brassica napus L.) lines. Plant. Genome 2018, 11, 170100. [Google Scholar] [CrossRef] [PubMed]
- Genesys. Available online: https://www.genesys-pgr.org/10.18730/5PNPV (accessed on 21 December 2020).
- Qureshi, A.S.; Ertebo, T.; Mehansiwala, M. Prospects of alternative copping systems for salt affected soils in Ethiopia. J. Soil Sci. Environ. Manag. 2018, 9, 98–107. [Google Scholar] [CrossRef]
- Genesys. Available online: https://www.genesys-pgr.org/10.18730/8VSA4 (accessed on 21 December 2020).
- Materne, M.; Murden, S.; Holding, B.; Krishna, B. Lentil Variety Evaluation. 2000. Available online: https://www.farmtrials.com.au/trial/13569 (accessed on 21 December 2020).
- Rao, A.Q.; ud Din, S.; Akhtar, S.; Sarwar, M.B.; Ahmed, M.; Rashid, B.; Khan, M.A.U.; Qaisar, U.; Shahid, A.A.; Nasir, I.A.; et al. Genomics of salinity tolerance in plants. Plant. Genom. 2016, 273. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, W.; Tong, W.; He, Q.; Yoon, M.Y.; Li, F.P.; Choi, B.; Heo, E.B.; Kim, K.W.; Park, Y.J. A genome-wide association study reveals candidate genes related to salt tolerance in rice (Oryza sativa) at the germination stage. Int. J. Mol. Sci. 2018, 19, 3145. [Google Scholar] [CrossRef] [PubMed]
- Shohan, M.; Sinha, S.; Nabila, F.H.; Dastidar, S.G.; Seraj, Z.I. HKT1;5 Transporter gene expression and association of amino acid substitutions with salt tolerance across rice genotypes. Front.Plant. Sci. 2019, 10, 1420. [Google Scholar] [CrossRef]
- Hazzouri, K.M.; Khraiwesh, B.; Amiri, K.; Pauli, D.; Blake, T.; Shahid, M.; Mullath, S.K.; Nelson, D.; Mansour, A.L.; Salehi-Ashtiani, K.; et al. Mapping of HKT1;5 gene in barley using gwas approach and its implication in salt tolerance mechanism. Front. Plant. Sci. 2018, 9, 156. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filtering Step | Targeted-GBS (tGBS) Method | Transcriptome-Based GBS (GBS-t) Method |
|---|---|---|
| Depth 5 | 2,043,680 | 1,614,141 |
| Maximum missing 80 and Q30 | 457,692 | 90,493 |
| Heterozygosity 0.2 | 450,927 | 85,641 |
| Minor allelic frequency 0.05 | 57,344 | 53,186 |
| Final analysis | 56,349 | 52,471 |
| Position | GBS Method | Alleles | Tolerant | Intolerant | ||
|---|---|---|---|---|---|---|
| Favorable Allele | No. of Accessions | Favorable Allele | No. of Accessions | |||
| 392,560,939 | tGBS | A/G | A | 28 | G | 34 |
| 392,619,081 | tGBS | C/G | C | 26 | G | 35 |
| 392,619,090 | tGBS | C/T | T | 26 | C | 35 |
| 392,619,094 | tGBS | A/T | A | 26 | T | 35 |
| 392,619,099 | tGBS | A/G | A | 26 | G | 35 |
| 392,619,165 | tGBS | A/C | C | 28 | A | 35 |
| 392,690,695 | tGBS | C/T | T | 25 | C | 33 |
| 392,690,695 | GBS-t | C/T | T | 31 | C | 27 |
| 392,690,876 | GBS-t | A/G | G | 31 | A | 29 |
| 392,692,372 | GBS-t | T/G | G | 31 | T | 31 |
| 392,692,397 | GBS-t | A/G | G | 29 | A | 32 |
| 392,695,004 | GBS-t | A/G | G | 31 | A | 29 |
| 392,695,044 | GBS-t | C/T | T | 32 | C | 32 |
| 392,695,091 | tGBS | A/G | A | 23 | G | 35 |
| 392,695,091 | GBS-t | A/G | A | 32 | G | 32 |
| 392,695,192 | tGBS | C/T | T | 23 | C | 35 |
| 392,697,101 | GBS-t | T/C | C | 32 | T | 32 |
| 392,697,852 | GBS-t | T/A | A | 30 | T | 32 |
| 392,697,951 | GBS-t | T/C | C | 29 | T | 32 |
| 392,699,727 | GBS-t | T/C | C | 32 | T | 31 |
| 392,700,661 | GBS-t | C/A | A | 31 | C | 31 |
| 392,758,269 | tGBS | C/T | C | 26 | T | 33 |
| 392,767,939 | GBS-t | C/A | A | 28 | C | 30 |
| 393,426,328 | tGBS | G/T | T | 26 | G | 35 |
| 393,426,332 | tGBS | C/T | T | 22 | C | 35 |
| 393,427,373 | GBS-t | C/T | T | 31 | C | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dissanayake, R.; Cogan, N.O.I.; Smith, K.F.; Kaur, S. Application of Genomics to Understand Salt Tolerance in Lentil. Genes 2021, 12, 332. https://doi.org/10.3390/genes12030332
Dissanayake R, Cogan NOI, Smith KF, Kaur S. Application of Genomics to Understand Salt Tolerance in Lentil. Genes. 2021; 12(3):332. https://doi.org/10.3390/genes12030332
Chicago/Turabian StyleDissanayake, Ruwani, Noel O.I. Cogan, Kevin F. Smith, and Sukhjiwan Kaur. 2021. "Application of Genomics to Understand Salt Tolerance in Lentil" Genes 12, no. 3: 332. https://doi.org/10.3390/genes12030332
APA StyleDissanayake, R., Cogan, N. O. I., Smith, K. F., & Kaur, S. (2021). Application of Genomics to Understand Salt Tolerance in Lentil. Genes, 12(3), 332. https://doi.org/10.3390/genes12030332