
G2PMineR: A Genome to Phenome Literature Review Approach




Abstract
1. Introduction
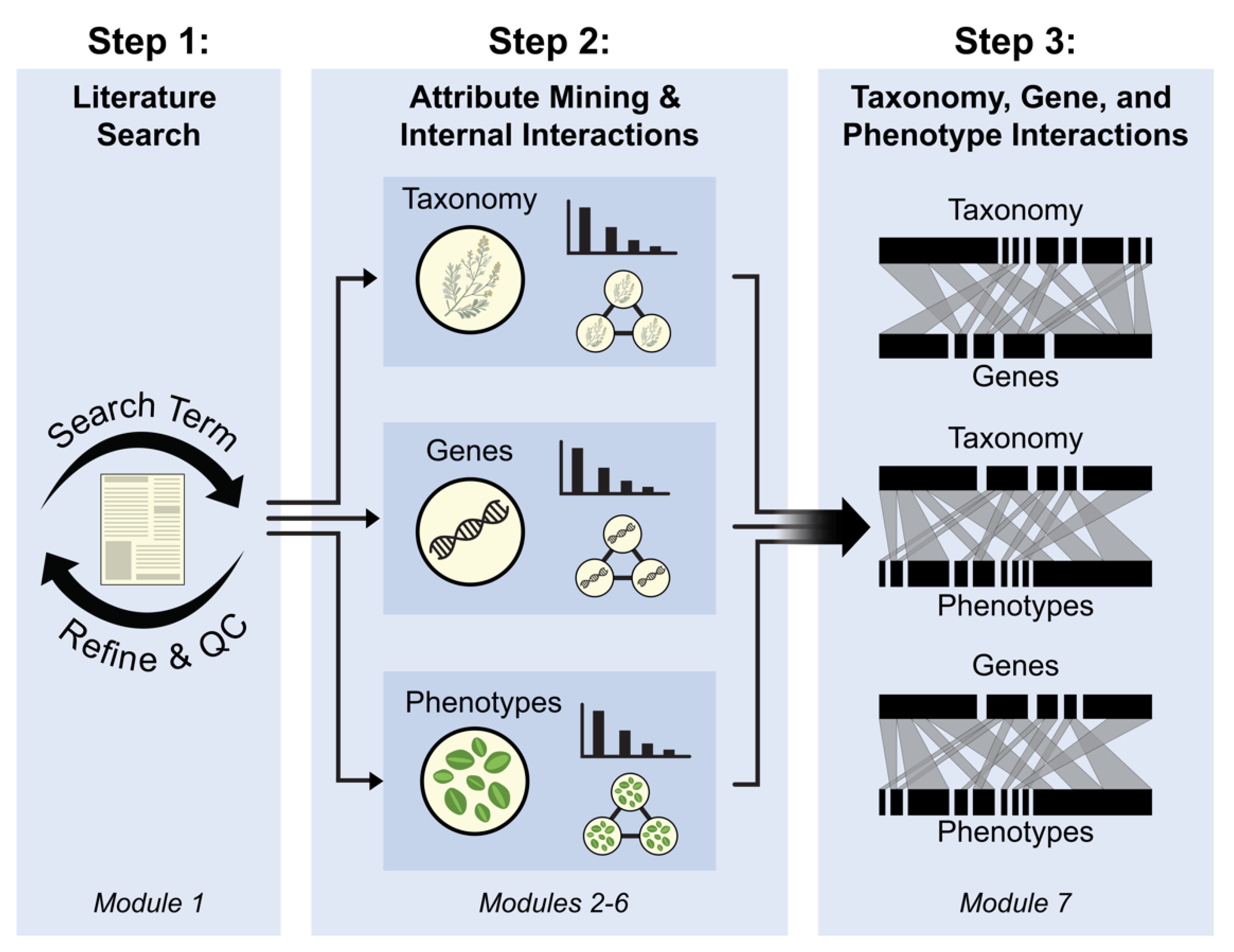
2. Materials and Methods
2.1. General Structure
2.2. Input Data
2.3. Step 1: Literature Search
Module 1: Conduct Literature Search and Assess its Efficiency (Optional)
2.4. Step 2 Attribute Mining and Internal Interactions
2.4.1. Module 2: Mining Taxonomy (Ta)
2.4.2. Module 3: Mining Genes (G)
2.4.3. Module 4: Mining Phenotypes (P)
2.4.4. Module 5: Summarizing and Inferring Consensus of Genes, Taxonomy, and Phenotypes Data
2.4.5. Module 6: Internal Network Analyses for Ta, G, and P Data
2.5. Step 3: Linking Ta, G, and P Interactions
Module 7: Constructing Bipartite Graphs
2.6. Operating System and R Versioning
3. Results
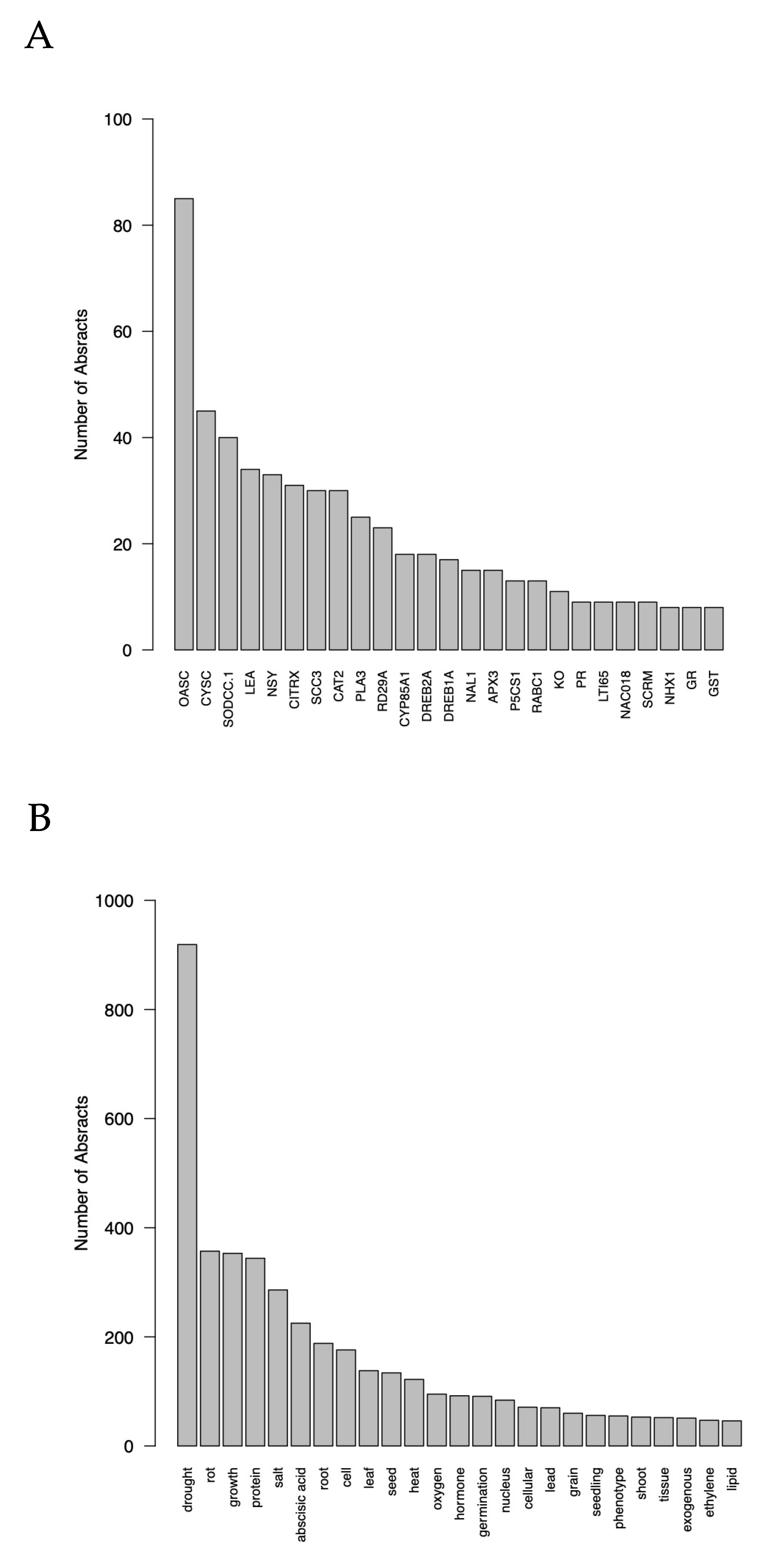
3.1. Step 1: Literature Search Results
Module 1: Conduct Literature Search and Assess its Efficiency Results
3.2. Step 2 Attribute Mining and Internal Interactions Results
3.2.1. Modules 2–4: Mining Results
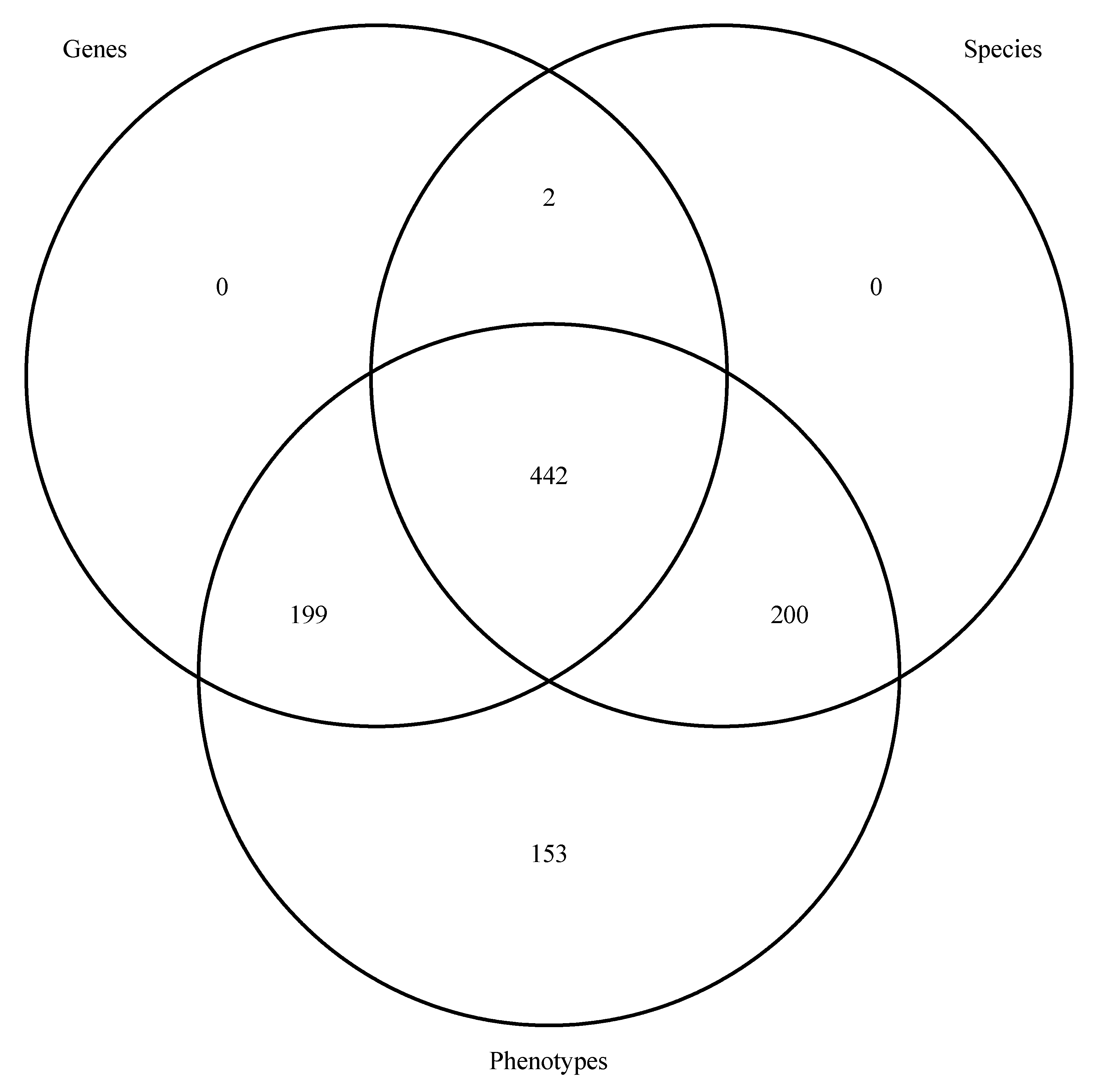
3.2.2. Module 5: Summarizing and Inferring Consensus of Ta, G, and P Term Matches Results
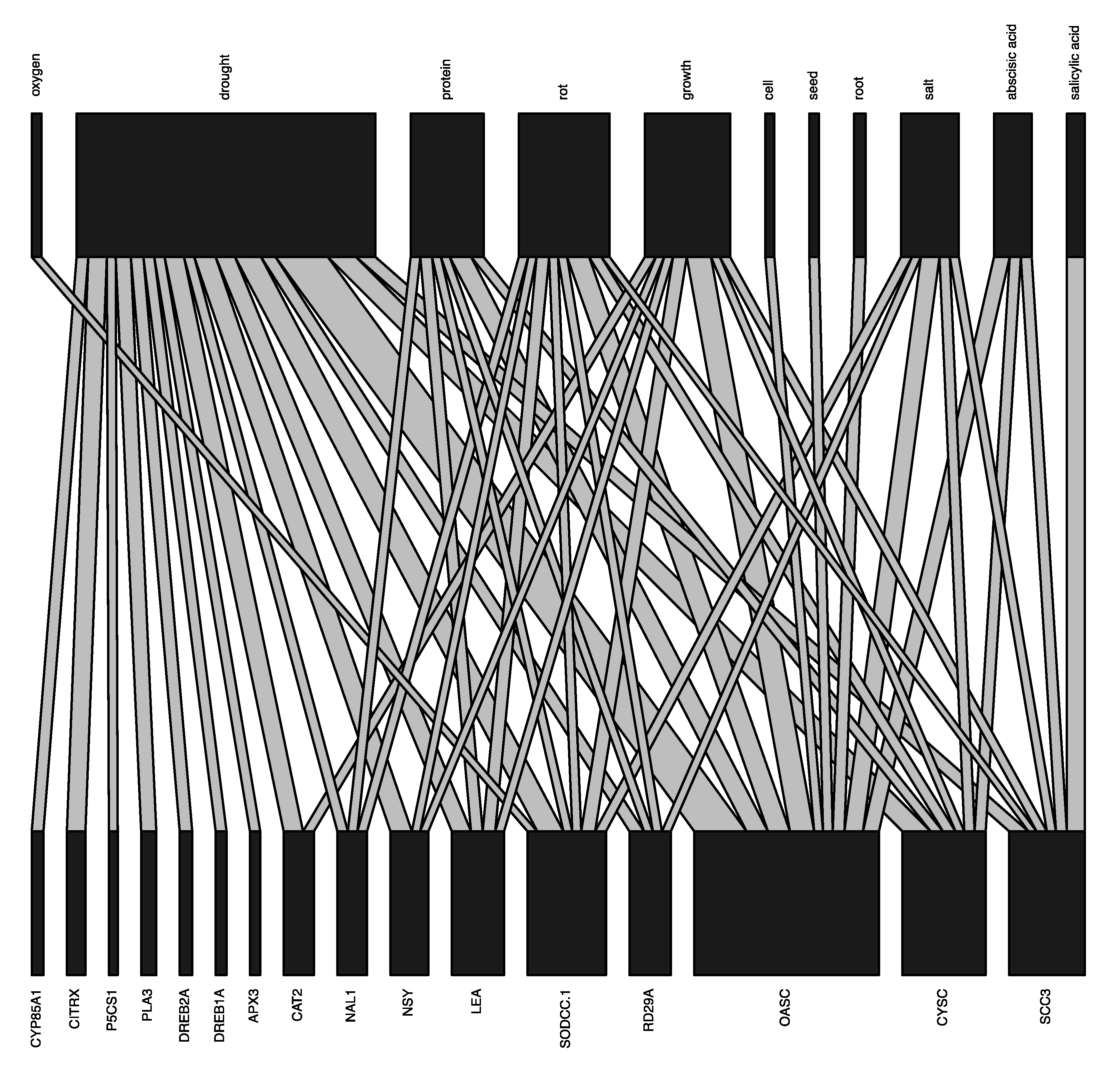
3.2.3. Module 6: Internal Network Analyses for G, Ta and P Data Results
3.3. Step 3: Linking Ta, G, and P Interactions Results
Module 7: Constructing Bipartite Graphs Results
4. Discussion
4.1. Our G2P Analysis Produced Results Aligned with Current Knowledge on Plants Drought Tolerance
4.2. G2PMineR Is Applicable to Studying G2P in Plants, Animals, and Fungi
4.3. From Literature Review to Hypothesis Testing
4.4. G2PMineR Was Designed with Diverse Users in Mind
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwon, J.M.; Goate, A.M. The candidate gene approach. Alcohol Res. Health 2000, 24, 164–168. [Google Scholar] [PubMed]
- Moore, S.R. Commentary: What Is the Case for Candidate Gene Approaches in the Era of High-Throughput Genomics? A Response to Border and Keller. J. Child Psychol. Psychiatry 2017, 58, 331–334. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Idaho GEM3 Genes by Environment. Available online: https://www.idahogem3.org/ (accessed on 18 December 2020).
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The Power and Promise of Population Genomics: From Genotyping to Genome Typing. Nat. Rev. Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef]
- Ellegren, H. Genome Sequencing and Population Genomics in Non-Model Organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef]
- Tao, Y.; Cai, C.; Cohen, W.W.; Lu, X. From genome to phenome: Predicting multiple cancer phenotypes based on somatic genomic alterations via the genomic impact transformer. In Biocomputing 2020; World Scientific: Singapore, 2019; pp. 79–90. ISBN 9789811215629. [Google Scholar]
- London, S.J.; Romieu, I. Gene by Environment Interaction in Asthma. Annu. Rev. Public Health 2009, 30, 55–80. [Google Scholar] [CrossRef]
- Lendenmann, M.H.; Croll, D.; Palma-Guerrero, J.; Stewart, E.L.; McDonald, B.A. QTL Mapping of Temperature Sensitivity Reveals Candidate Genes for Thermal Adaptation and Growth Morphology in the Plant Pathogenic Fungus Zymoseptoria Tritici. Heredity 2016, 116, 384–394. [Google Scholar] [CrossRef]
- Russell, J.J.; Theriot, J.A.; Sood, P.; Marshall, W.F.; Landweber, L.F.; Fritz-Laylin, L.; Polka, J.K.; Oliferenko, S.; Gerbich, T.; Gladfelter, A.; et al. Non-Model Model Organisms. BMC Biol. 2017, 15, 55. [Google Scholar] [CrossRef]
- Galla, S.J.; Forsdick, N.J.; Brown, L.; Hoeppner, M.P.; Knapp, M.; Maloney, R.F.; Moraga, R.; Santure, A.W.; Steeves, T.E. Reference Genomes from Distantly Related Species Can Be Used for Discovery of Single Nucleotide Polymorphisms to Inform Conservation Management. Genes 2019, 10, 9. [Google Scholar] [CrossRef]
- Burnett, K.G.; Durica, D.S.; Mykles, D.L.; Stillman, J.H.; Schmidt, C. Recommendations for Advancing Genome to Phenome Research in Non-Model Organisms. Integr. Comp. Biol. 2020, 60, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Zargar, S.M.; Raatz, B.; Sonah, H.; Bhat, J.A.; Dar, Z.A.; Agrawal, G.K.; Rakwal, R. Recent Advances in Molecular Marker Techniques: Insight into QTL Mapping, GWAS and Genomic Selection in Plants. J. Crop Sci. Biotechnol. 2015, 18, 293–308. [Google Scholar] [CrossRef]
- Van Egmond, M.E.; Lugtenberg, C.H.A.; Brouwer, O.F.; Contarino, M.F.; Fung, V.S.C.; Heiner-Fokkema, M.R.; van Hilten, J.J.; van der Hout, A.H.; Peall, K.J.; Sinke, R.J.; et al. A Post Hoc Study on Gene Panel Analysis for the Diagnosis of Dystonia. Mov. Disord. 2017, 32, 569–575. [Google Scholar] [CrossRef]
- Zhu, M.; Zhao, S. Candidate Gene Identification Approach: Progress and Challenges. Int. J. Biol. Sci. 2007, 3, 420–427. [Google Scholar] [CrossRef]
- Border, R.; Keller, M.C. Commentary: Fundamental Problems with Candidate Gene-by-Environment Interaction Studies—Reflections on Moore and Thoemmes. J. Child Psychol. Psychiatry 2017, 58, 328–330. [Google Scholar] [CrossRef]
- Bakshi, R.K.; Kaur, N.; Kaur, R.; Kaur, G. Opinion Mining and Sentiment Analysis. In Proceedings of the 2016 3rd International Conference on Computing for Sustainable Global Development (INDIACom), New Delhi, India, 16–18 March 2016; pp. 452–455. [Google Scholar]
- Aria, M.; Cuccurullo, C. Bibliometrix: An R-Tool for Comprehensive Science Mapping Analysis. J. Informetr. 2017, 11, 959–975. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2019. Available online: https://www.R-project.org/ (accessed on 2 February 2021).
- Wickham, H.; Hester, J.; Chang, W. Some namespace and vignette code extracted from base. In Devtools: Tools to Make Developing R Packages Easier; R Studio Team: Boston, MA, USA, 2020. [Google Scholar]
- Roberts, R.J. PubMed Central: The GenBank of the Published Literature. Proc. Natl. Acad. Sci. USA 2001, 98, 381–382. [Google Scholar] [CrossRef]
- Burnham, J.F. Scopus Database: A Review. Biomed. Digit. Libr. 2006, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Harzing, A.-W.; Alakangas, S. Google Scholar, Scopus and the Web of Science: A Longitudinal and Cross-Disciplinary Comparison. Scientometrics 2016, 106, 787–804. [Google Scholar] [CrossRef]
- Kovalchik, S. RISmed: Download Content from NCBI Databases. R Package Version 2.2. 2020. Available online: https://CRAN.R-project.org/package=RISmed (accessed on 2 February 2021).
- Fantini, D. easyPubMed: Search and Retrieve Scientific Publication Records from PubMed. R Package Version 2.13. 2019. Available online: https://CRAN.R-project.org/package=easyPubMed (accessed on 2 February 2021).
- Selivanov, D.; Bickel, M.; Wang, Q. text2vec: Modern Text Mining Framework for R. R Package Version 0.6. 2020. Available online: https://CRAN.R-project.org/package=text2vec (accessed on 2 February 2021).
- Epskamp, S.; Cramer, A.O.J.; Waldorp, L.J.; Schmittmann, V.D.; Borsboom, D. qgraph: Network Visualizations of Relationships in Psychometric Data. J. Stat. Softw. 2012, 48, 1–18. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Global Biodiversity Information Facility. Gbif Memo. Underst. 2010. [CrossRef]
- Chamberlain, S.; Szocs, E. taxize—Taxonomic search and retrieval in R. F1000Research. 2013. Available online: http://f1000research.com/articles/2-191/v2 (accessed on 2 February 2021).
- Cayuela, L.; Macarro, I.; Stein, A.; Oksanen, J. Taxonstand: Taxonomic Standardization of Plant Species Names. Methods Ecol. Evol. 2019, 3, 1078–1083. [Google Scholar] [CrossRef]
- Bairoch, A.; Boeckmann, B. The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 1991, 19, 2247–2249. [Google Scholar] [CrossRef] [PubMed]
- Missouri Botanical Gardens. Available online: http://www.mobot.org/MOBOT/Research/APweb/top/glossarya_h.html (accessed on 2 February 2021).
- Collins, A.; Speer, B.; Waggoner, B.; Whitney, C.; Rieboldt, S. UC Museum of Paleontology Glossary: Zoology. Available online: https://ucmp.berkeley.edu/glossary/augloss.html (accessed on 21 December 2020).
- Ellis, D. Glossary of Mycological Terms | Mycology Online. Available online: https://mycology.adelaide.edu.au/glossary/ (accessed on 21 December 2020).
- Chen, H. VennDiagram: Generate High-Resolution Venn and Euler Plots. R Package Version 1.6.20. 2018. Available online: https://CRAN.R-project.org/package=VennDiagram (accessed on 2 February 2021).
- Dormann, C.F.; Gruber, B.; Freund, J. Introducing the Bipartite Package: Analysing Ecological Networks. Interaction 2008, 1, 0.2413793. [Google Scholar]
- Estravis-Barcala, M.; Mattera, M.G.; Soliani, C.; Bellora, N.; Opgenoorth, L.; Heer, K.; Arana, M.V. Molecular Bases of Responses to Abiotic Stress in Trees. J. Exp. Bot. 2020, 71, 3765–3779. [Google Scholar] [CrossRef]
- Jenks, M.A.; Hasegawa, P.M. Plant Abiotic Stress; Blackwell Publishing: Hoboken, NJ, USA, 2005. [Google Scholar]
- Haak, D.C.; Fukao, T.; Grene, R.; Hua, Z.; Ivanov, R.; Perrella, G.; Li, S. Multilevel Regulation of Abiotic Stress Responses in Plants. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Striberny, B.; Melton, A.E.; Schwacke, R.; Krause, K.; Fischer, K.; Goertzen, L.R.; Rashotte, A.M. Cytokinin Response Factor 5 Has Transcriptional Activity Governed by Its C-terminal Domain. Plant Signal. Behav. 2017, 12, e1276684. [Google Scholar] [CrossRef][Green Version]
- Menezes-Benavente, L.; Teixeira, F.K.; Alvim Kamei, C.L.; Margis-Pinheiro, M. Salt Stress Induces Altered Expression of Genes Encoding Antioxidant Enzymes in Seedlings of a Brazilian Indica Rice (Oryza sativa L.). Plant Sci. 2004, 166, 323–331. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group 1 | Group 2 |
|---|---|
| Drought * | Tolerance * |
| Tolerance * | Drought * |
| Gene * | Stress * |
| Stress * | Plant * |
| Plant * | Gene * |
| Expression * | Expression * |
| Response * | Response * |
| Under * | Transgenic |
| Study * | Protein |
| Analysis | Result |
| Abiotic | |
| Role | |
| Study * | |
| Under * | |
| Acid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojahn, J.M.A.; Galla, S.J.; Melton, A.E.; Buerki, S. G2PMineR: A Genome to Phenome Literature Review Approach. Genes 2021, 12, 293. https://doi.org/10.3390/genes12020293
Wojahn JMA, Galla SJ, Melton AE, Buerki S. G2PMineR: A Genome to Phenome Literature Review Approach. Genes. 2021; 12(2):293. https://doi.org/10.3390/genes12020293
Chicago/Turabian StyleWojahn, John M. A., Stephanie J. Galla, Anthony E. Melton, and Sven Buerki. 2021. "G2PMineR: A Genome to Phenome Literature Review Approach" Genes 12, no. 2: 293. https://doi.org/10.3390/genes12020293
APA StyleWojahn, J. M. A., Galla, S. J., Melton, A. E., & Buerki, S. (2021). G2PMineR: A Genome to Phenome Literature Review Approach. Genes, 12(2), 293. https://doi.org/10.3390/genes12020293