
Comparative Mitogenomics in Hyalella (Amphipoda: Crustacea)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction, Mitogenome Assembly and Annotation
2.3. Composition Analyses and Gene Order
2.4. Analyses of the Secondary Structure of tRNAs
2.5. Phylogenetic Inference and Selection Analyses
3. Results
3.1. Genome Organization
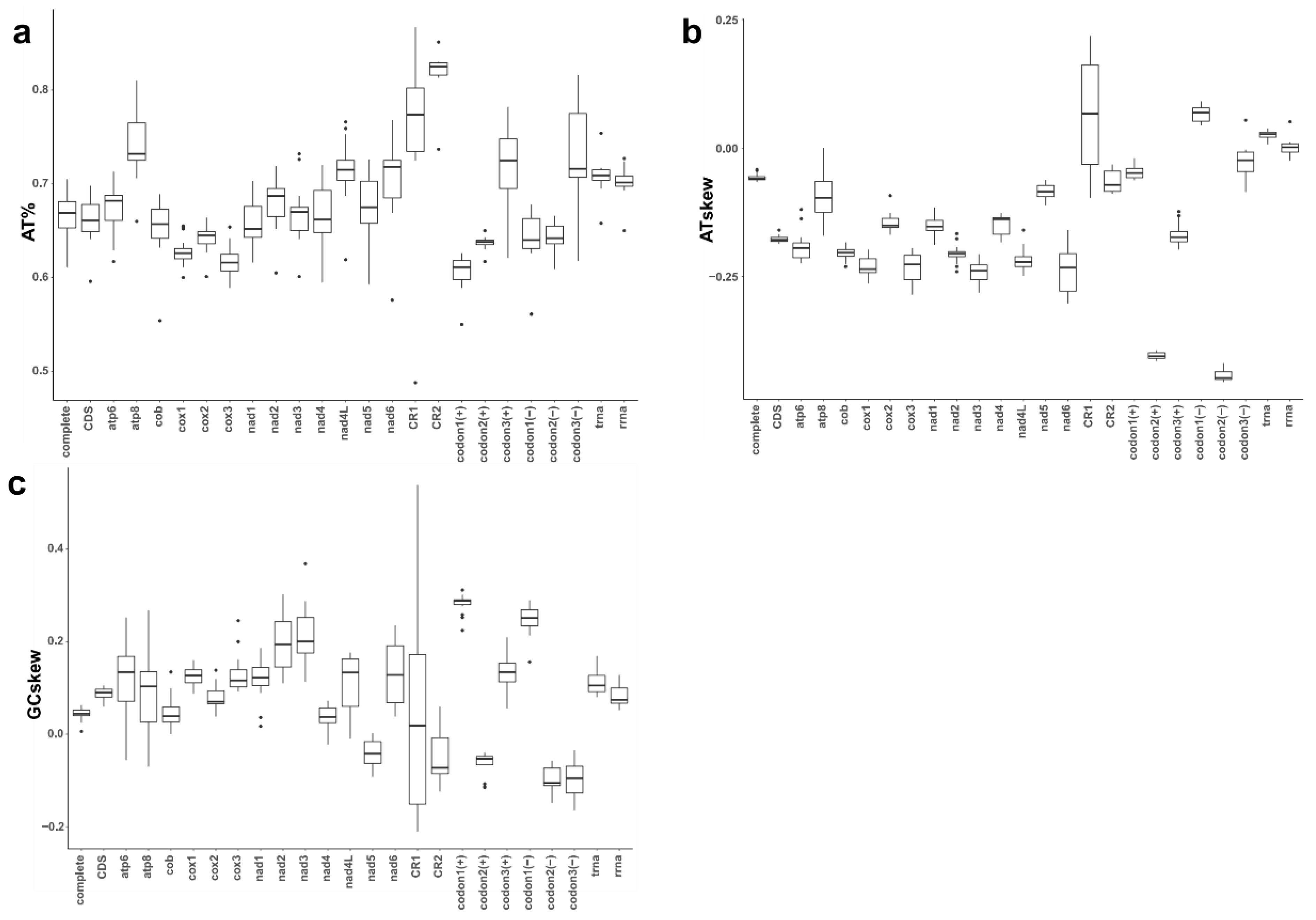
3.2. Base Composition and AT- and GC-Skews
3.3. Start and Stop Codons, Amino Acid Composition and Codon Usage
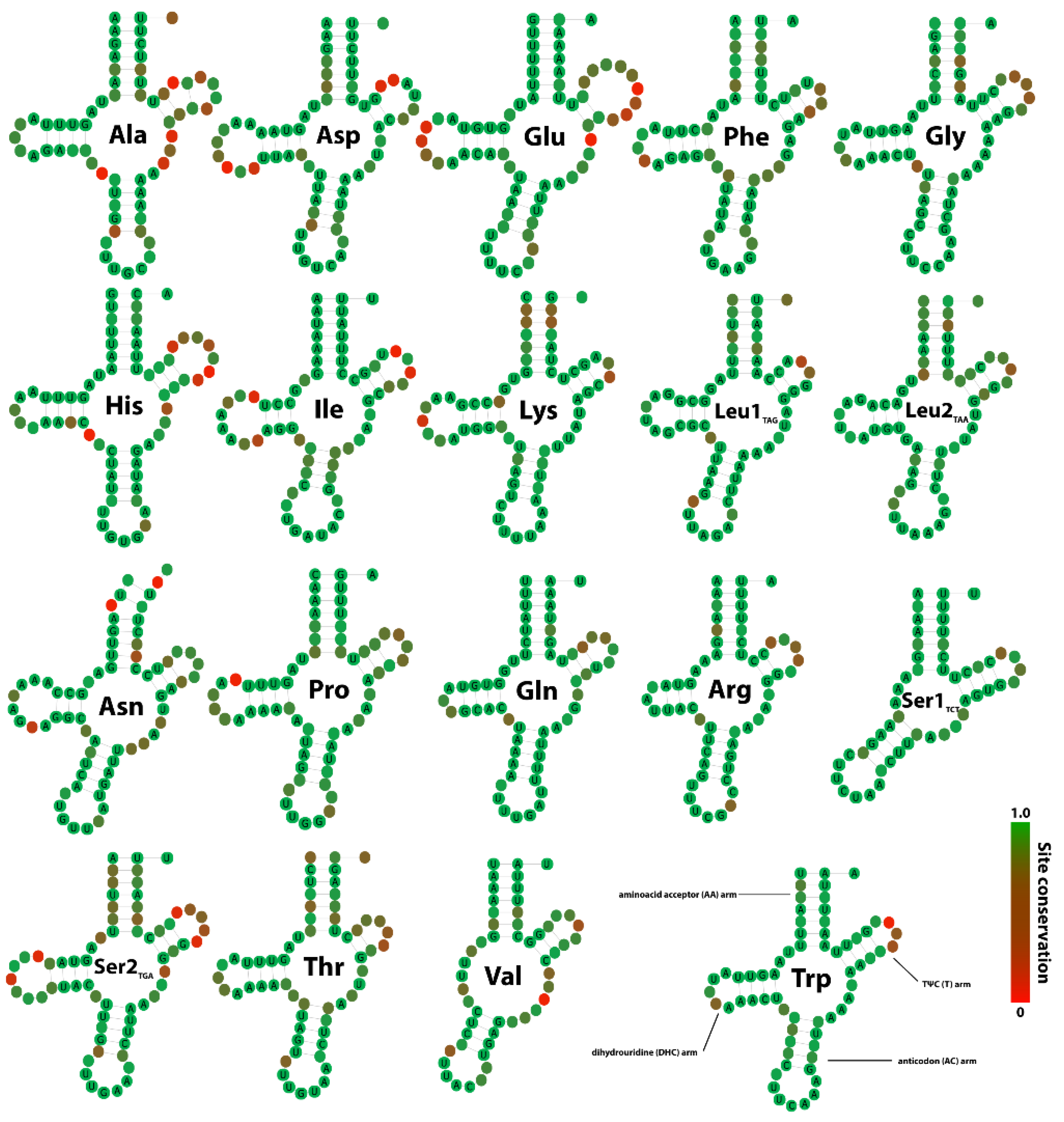
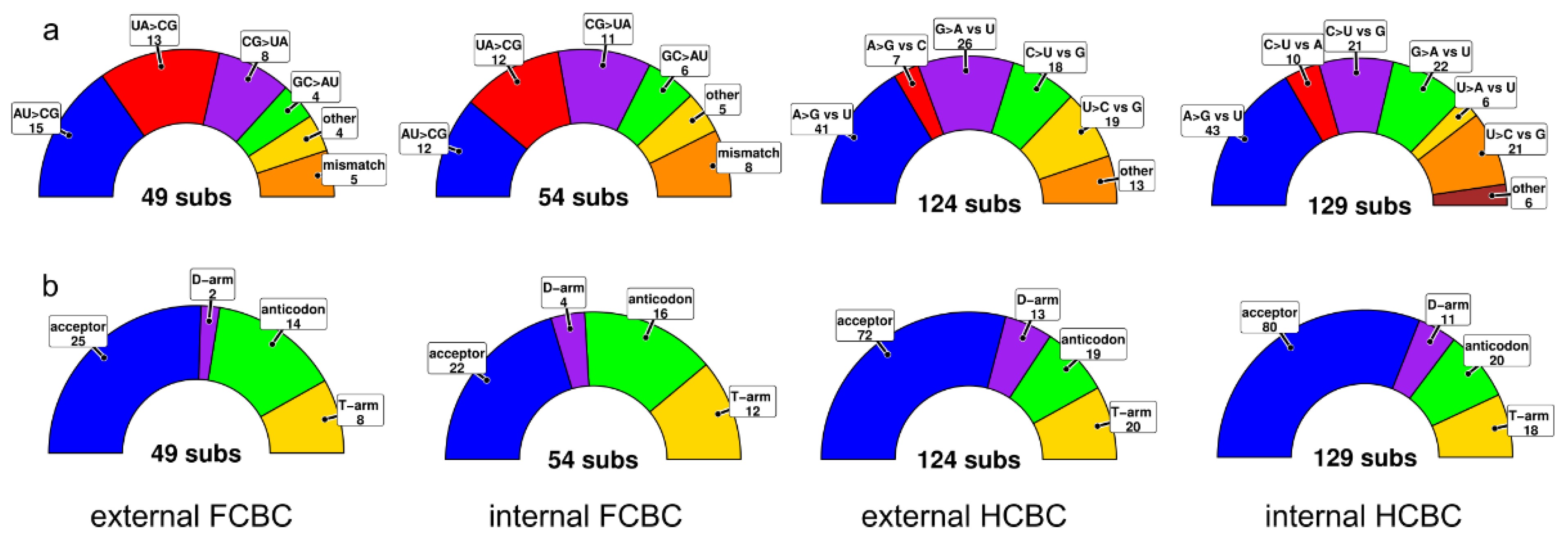
3.4. tRNA Secondary Structures and Compensatory Mutations
3.5. Positive Selection Analyses in Protein-Coding Genes
4. Discussion
4.1. Hyalella mtDNA Sequence Features
4.2. Mitochondrial Gene Arrangement in Hyalella
4.3. Secondary Structures and Compensatory Base Changes in tRNAs
4.4. Selection in Protein-Coding Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bauzà-Ribot, M.M.; Juan, C.; Nardi, F.; Oromí, P.; Pons, J.; Jaume, D. Mitogenomic Phylogenetic Analysis Supports Continental-Scale Vicariance in Subterranean Thalassoid Crustaceans. Curr. Biol. 2012, 22, 2069–2074. [Google Scholar] [CrossRef]
- Pons, J.; Bauzà-Ribot, M.M.; Jaume, D.; Juan, C. Next-generation sequencing, phylogenetic signal and comparative mitogenomic analyses in Metacrangonyctidae (Amphipoda: Crustacea). BMC Genom. 2014, 15, 566. [Google Scholar] [CrossRef]
- Romanova, E.V.; Aleoshin, V.V.; Kamaltynov, R.M.; Mikhailov, K.V.; Logacheva, M.D.; Sirotinina, E.A.; Gornov, A.Y.; Anikin, A.S.; Sherbakov, D.Y. Evolution of mitochondrial genomes in Baikalian amphipods. BMC Genom. 2016, 17, 1016–1306. [Google Scholar] [CrossRef] [PubMed]
- Stokkan, M.; Jurado-Rivera, J.A.; Oromí, P.; Juan, C.; Jaume, D.; Pons, J. Species delimitation and mitogenome phylogenetics in the subterranean genus Pseudoniphargus (Crustacea: Amphipoda). Mol. Phylogenet. Evol. 2018, 127, 988–999. [Google Scholar] [CrossRef]
- Waeschenbach, A.; Telford, M.J.; Porter, J.S.; Littlewood, D.T.J. The complete mitochondrial genome of Flustrellidra hispida and the phylogenetic position of Bryozoa among the Metazoa. Mol. Phylogenet. Evol. 2006, 40, 195–207. [Google Scholar] [CrossRef]
- Juan, C.; Jurado-Rivera, J.A.; Moreno, E.; Wolff, C.; Jaume, D.; Pons, J. The mitogenome of the amphipod Hyalella lucifugax (Crustacea) and its phylogenetic placement. Mitochondrial DNA Part B 2016, 1, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Tamura, K.; Aotsuka, T. Replication Origin of Mitochondrial DNA in Insects. Genetics 2005, 171, 1695–1705. [Google Scholar] [CrossRef]
- Fernández-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and Transcription of Mammalian Mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Pons, J.; Bover, P.; Bidegaray-Batista, L.; Arnedo, M.A. Armless mitochondrial tRNAs conserved for over 30 millions of years in spiders. BMC Genom. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Montelli, S.; Peruffo, A.; Patarnello, T.; Cozzi, B.; Negrisolo, E. Back to Water: Signature of Adaptive Evolution in Cetacean Mitochondrial tRNAs. PLoS ONE 2016, 11, e0158129. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, Y.; Ota, H.; Nishida, M.; Ozawa, T. Gene rearrangements in snake mitochondrial genomes: Highly concerted evolution of control-region-like sequences duplicated and inserted into a tRNA gene cluster. Mol. Biol. Evol. 1996, 13, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Stokkan, M.; Jurado-Rivera, J.A.; Juan, C.; Jaume, D.; Pons, J. Mitochondrial genome rearrangements at low taxonomic levels: Three distinct mitogenome gene orders in the genus Pseudoniphargus (Crustacea: Amphipoda). Mitochondrial DNA Part A 2015, 27, 3579–3589. [Google Scholar] [CrossRef]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Collins, T.M.; Stanton, D.; Daehler, L.L.; Brown, W.M. Deducing the pattern of arthropod phytogeny from mitochondrial DNA rearrangements. Nature 1995, 376, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Castellana, S.; Vicario, S.; Saccone, C. Evolutionary Patterns of the Mitochondrial Genome in Metazoa: Exploring the Role of Mutation and Selection in Mitochondrial Protein–Coding Genes. Genome Biol. Evol. 2011, 3, 1067–1079. [Google Scholar] [CrossRef]
- Allio, R.; Donega, S.; Galtier, N.; Nabholz, B. Large Variation in the Ratio of Mitochondrial to Nuclear Mutation Rate across Animals: Implications for Genetic Diversity and the Use of Mitochondrial DNA as a Molecular Marker. Mol. Biol. Evol. 2017, 34, 2762–2772. [Google Scholar] [CrossRef]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for Multiple Reversals of Asymmetric Mutational Constraints during the Evolution of the Mitochondrial Genome of Metazoa, and Consequences for Phylogenetic Inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, T.; Nikaido, M.; Tsuchiya, K.; Segawa, S.; Hasegawa, M.; Okada, N. Extensive mitochondrial gene arrangements in coleoid Cephalopoda and their phylogenetic implications. Mol. Phylogenet. Evol. 2006, 38, 648–658. [Google Scholar] [CrossRef]
- Yan, J.; Li, H.; Zhou, K. Evolution of the Mitochondrial Genome in Snakes: Gene Rearrangements and Phylogenetic Relationships. BMC Genom. 2008, 9, 569. [Google Scholar] [CrossRef]
- Dowton, M.; Cameron, S.L.; Dowavic, J.I.; Austin, A.D.; Whiting, M.F. Characterization of 67 Mitochondrial tRNA Gene Rearrangements in the Hymenoptera Suggests That Mitochondrial tRNA Gene Position Is Selectively Neutral. Mol. Biol. Evol. 2009, 26, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Arndt, A.; Smith, M.J. Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria. Mol. Biol. Evol. 1998, 15, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Kurabayashi, A.; Yoshikawa, N.; Sato, N.; Hayashi, Y.; Oumi, S.; Fujii, T.; Sumida, M. Complete mitochondrial DNA sequence of the endangered frog Odorrana ishikawae (family Ranidae) and unexpected diversity of mt gene arrangements in ranids. Mol. Phylogenet. Evol. 2010, 56, 543–553. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Yanase, T.; Tsuda, T.; Noda, H. Species-specific mitochondrial gene rearrangements in biting midges and vector species identification. Med. Vet. Entomol. 2009, 23, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2011, 40, 2833–2845. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.D.; Kondrashov, F.A. Mechanisms and convergence of compensatory evolution in mammalian mitochondrial tRNAs. Nat. Genet. 2004, 36, 1207–1212. [Google Scholar] [CrossRef]
- Meer, M.V.; Kondrashov, A.S.; Artzy-Randrup, Y.; Kondrashov, F.A. Compensatory evolution in mitochondrial tRNAs navigates valleys of low fitness. Nature 2010, 464, 279–282. [Google Scholar] [CrossRef]
- Naumenko, S.A.; Logacheva, M.D.; Popova, N.V.; Klepikova, A.V.; Penin, A.A.; Bazykin, G.A.; Etingova, A.E.; Mugue, N.S.; Kondrashov, A.S.; Yampolsky, L.Y. Transcriptome-based phylogeny of endemic Lake Baikal amphipod species flock: Fast speciation accompanied by frequent episodes of positive selection. Mol. Ecol. 2017, 26, 536–553. [Google Scholar] [CrossRef]
- Guo, H.; Yang, H.; Tao, Y.; Tang, D.; Wu, Q.; Wang, Z.; Tang, B. Mitochondrial OXPHOS genes provides insights into genetics basis of hypoxia adaptation in anchialine cave shrimps. Genes Genom. 2018, 40, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, X.; Sun, L.; Bai, Y.; Zhang, D.; Tang, B. Evolution of mitochondrial energy metabolism genes associated with hydrothermal vent adaption of Alvinocaridid shrimps. Genes Genom. 2017, 39, 1367–1376. [Google Scholar] [CrossRef]
- Grosso, L.E.; Peralta, M. Anfípodos de agua dulce sudamericanos. Revisión del género Hyalella Smith. I. Acta Zool. Lilloana 1999, 45, 79–98. [Google Scholar]
- Jurado-Rivera, J.A.; Zapelloni, F.; Pons, J.; Juan, C.; Jaume, D. Morphological and molecular species boundaries in the Hyalella species flock of Lake Titicaca (Crustacea: Amphipoda). Contrib. Zool. 2020, 89, 353–372. [Google Scholar] [CrossRef]
- Adamowicz, S.J.; Marinone, M.C.; Marque, S.M.; Martin, J.W.; Allen, D.C.; Pyle, M.N.; Ríos, P.D.L.; Sobel, C.N.; Ibañez, C.; Pinto, J.; et al. The Hyalella (Crustacea: Amphipoda) species cloud of the ancient Lake Titicaca originated from multiple colonizations. Mol. Phylogenet. Evol. 2018, 125, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Zapelloni, F.; Pons, J.; Jurado-Rivera, J.A.; Jaume, D.; Juan, C. Phylogenomics of the Hyalella amphipod species-flock of the Andean Altiplano. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Schomaker-Bastos, A.; Prosdocimi, F. MitoMaker: A pipeline for automatic assembly and annotation of animal mitochondria using raw NGS data. Preprints 2018, 1–10. [Google Scholar] [CrossRef]
- Geneious. Bioinformatics Software for Sequence Data Analysis. Available online: https://www.geneious.com/ (accessed on 10 September 2020).
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Narakusumo, R.P.; Riedel, A.; Pons, J. Mitochondrial genomes of twelve species of hyperdiverse Trigonopterus weevils. PeerJ 2020, 8, e10017. [Google Scholar] [CrossRef]
- Caswell, T.A.; Droettboom, M.; Lee, A.; Hunter, J.; de Andrade, E.S.; Firing, E.; Hoffmann, T.; Klymak, J.; Stansby, D.; Varoquaux, N.; et al. matplotlib/matplotlib: REL: v3.3.2. Zenodo 2020. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 9783319242774. [Google Scholar]
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. Heatmaply: An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.; Kocher, T. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Vlahoviček, K. INCA: Synonymous codon usage analysis and clustering by means of self-organizing map. Bioinformatics 2004, 20, 2329–2330. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN: A program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Seibel, P.N.; Müller, T.; Dandekar, T.; Schultz, J.; Wolf, M. 4SALE—A tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinform. 2006, 7, 498. [Google Scholar] [CrossRef]
- Galili, T. Dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Baker, F.B. Stability of Two Hierarchical Grouping Techniques Case 1: Sensitivity to Data Errors. J. Am. Stat. Assoc. 1974, 69, 440. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Dunn, C.W. Phyutility: A phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 2008, 24, 715–716. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating Synonymous and Nonsynonymous Substitution Rates Under Realistic Evolutionary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Woolley, S.; Johnson, J.; Smith, M.J.; Crandall, K.A.; McClellan, D.A. TreeSAAP: Selection on Amino Acid Properties using phylogenetic trees. Bioinformatics 2003, 19, 671–672. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Z.; Xu, S.; Ren, W.; Zhou, K.; Yang, G. ‘Obesity’ is healthy for cetaceans? Evidence from pervasive positive selection in genes related to triacylglycerol metabolism. Sci. Rep. 2015, 5, 14187. [Google Scholar] [CrossRef]
- Kao, D.; Lai, A.G.; Stamataki, E.; Rošić, S.; Konstantinides, N.; Jarvis, E.; Di Donfrancesco, A.; Pouchkina-Stancheva, N.; Sémon, M.; Grillo, M.; et al. The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. eLife 2016, 5. [Google Scholar] [CrossRef]
- Yang, H.-M.; Song, J.-H.; Kim, M.-S.; Min, G.-S. The complete mitochondrial genomes of two talitrid amphipods, Platorchestia japonica and P. parapacifica (Crustacea, Amphipoda). Mitochondrial DNA Part B 2017, 2, 757–758. [Google Scholar] [CrossRef]
- Yang, J.-S.; Yang, W.-J. The complete mitochondrial genome sequence of the hydrothermal vent galatheid crab Shinkaia crosnieri (Crustacea: Decapoda: Anomura): A novel arrangement and incomplete tRNA suite. BMC Genom. 2008, 9, 257. [Google Scholar] [CrossRef]
- Bauzà-Ribot, M.M.; Jaume, D.; Juan, C.; Pons, J. The complete mitochondrial genome of the subterranean crustacean Metacrangonyx longipes (Amphipoda): A unique gene order and extremely short control region. Mitochondrial DNA 2009, 20, 88–99. [Google Scholar] [CrossRef]
- Hassanin, A. Phylogeny of Arthropoda inferred from mitochondrial sequences: Strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol. Phylogenet. Evol. 2006, 38, 100–116. [Google Scholar] [CrossRef]
- Wei, S.-J.; Shi, M.; Chen, X.-X.; Sharkey, M.J.; Van Achterberg, C.; Ye, G.-Y.; He, J.-H. New Views on Strand Asymmetry in Insect Mitochondrial Genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef]
- Boore, J.L.; Macey, J.; Medina, M. Sequencing and Comparing Whole Mitochondrial Genomes of Animals. Cellulases 2005, 395, 311–348. [Google Scholar] [CrossRef]
- Kilpert, F.; Podsiadlowski, L. The complete mitochondrial genome of the common sea slater, Ligia oceanica (Crustacea, Isopoda) bears a novel gene order and unusual control region features. BMC Genom. 2006, 7, 241. [Google Scholar] [CrossRef]
- Podsiadlowski, L.; Bartolomaeus, T. Major rearrangements characterize the mitochondrial genome of the isopod Idotea baltica (Crustacea: Peracarida). Mol. Phylogenet. Evol. 2006, 40, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, Y.; Lin, R.; Zhang, Y.; Hu, K.; Li, Y.; Huang, Z.; Peng, S.; Ding, J.; Geng, X.; et al. Mitochondrial genome characteristics of Somena scintillans (Lepidoptera: Erebidae) and comparation with other Noctuoidea insects. Genomics 2019, 111, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Rivera, J.A.; Álvarez, G.; Caro, J.A.; Juan, C.; Pons, J.; Jaume, D. Molecular systematics of Haploginglymus, a genus of subterranean amphipods endemic to the Iberian Peninsula (Amphipoda: Niphargidae). Contrib. Zool. 2017, 86, 239–260. [Google Scholar] [CrossRef]
- Hao, Y.-J.; Zou, Y.-L.; Ding, Y.-R.; Xu, W.-Y.; Yan, Z.-T.; Li, X.-D.; Fu, W.-B.; Li, T.-J.; Chen, B. Complete mitochondrial genomes of Anopheles stephensi and An. dirus and comparative evolutionary mitochondriomics of 50 mosquitoes. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Łukasik, P.; Chong, R.A.; Nazario, K.; Matsuura, Y.; Bublitz, D.A.C.; Campbell, M.A.; Meyer, M.C.; Van Leuven, J.T.; Pessacq, P.; Veloso, C.; et al. One Hundred Mitochondrial Genomes of Cicadas. J. Hered. 2019, 110, 247–256. [Google Scholar] [CrossRef]
- Pons, J.; Ribera, I.; Bertranpetit, J.; Balke, M. Nucleotide substitution rates for the full set of mitochondrial protein-coding genes in Coleoptera. Mol. Phylogenet. Evol. 2010, 56, 796–807. [Google Scholar] [CrossRef]
- Itzkovitz, S.; Alon, U. The genetic code is nearly optimal for allowing additional information within protein-coding sequences. Genome Res. 2007, 17, 405–412. [Google Scholar] [CrossRef]
- Creixell, P.; Schoof, E.M.; Tan, C.S.H.; Linding, R. Mutational properties of amino acid residues: Implications for evolvability of phosphorylatable residues. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2584–2593. [Google Scholar] [CrossRef]
- Rawlings, T.A.; Collins, T.M.; Bieler, R. Changing identities: tRNA duplication and remolding within animal mitochondrial genomes. Proc. Natl. Acad. Sci. USA 2003, 100, 15700–15705. [Google Scholar] [CrossRef]
- Krebes, L.; Bastrop, R. The mitogenome of Gammarus duebeni (Crustacea Amphipoda): A new gene order and non-neutral sequence evolution of tandem repeats in the control region. Comp. Biochem. Physiol. Part D Genom. Proteom. 2012, 7, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Patiño-Galindo, J.Á.; González-Candelas, F.; Pybus, O.G. The Effect of RNA Substitution Models on Viroid and RNA Virus Phylogenies. Genome Biol. Evol. 2018, 10, 657–666. [Google Scholar] [CrossRef]
- Kondrashov, F.A. Prediction of pathogenic mutations in mitochondrially encoded human tRNAs. Hum. Mol. Genet. 2005, 14, 2415–2419. [Google Scholar] [CrossRef][Green Version]
- Coleman, A.W. Is there a molecular key to the level of “biological species” in eukaryotes? A DNA guide. Mol. Phylogenet. Evol. 2009, 50, 197–203. [Google Scholar] [CrossRef]
- Meiklejohn, C.D.; Montooth, K.L.; Rand, D.M. Positive and negative selection on the mitochondrial genome. Trends Genet. 2007, 23, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wu, Y.; Ge, X.; Jakovlić, I.; Zhu, J.; Mahboob, S.; Al-Ghanim, K.A.; Al-Misned, F.; Fu, H. Disentangling the interplay of positive and negative selection forces that shaped mitochondrial genomes of Gammarus pisinnus and Gammarus lacustris. R. Soc. Open Sci. 2020, 7, 190669. [Google Scholar] [CrossRef]
- Pesole, G.; Gissi, C.; De Chirico, A.; Saccone, C. Nucleotide Substitution Rate of Mammalian Mitochondrial Genomes. J. Mol. Evol. 1999, 48, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Bazin, E.; Glémin, S.; Galtier, N. Population Size Does Not Influence Mitochondrial Genetic Diversity in Animals. Science 2006, 312, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.E.; Bell, M.A. Causes and consequences of recent freshwater invasions by saltwater animals. Trends Ecol. Evol. 1999, 14, 284–288. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapelloni, F.; Jurado-Rivera, J.A.; Jaume, D.; Juan, C.; Pons, J. Comparative Mitogenomics in Hyalella (Amphipoda: Crustacea). Genes 2021, 12, 292. https://doi.org/10.3390/genes12020292
Zapelloni F, Jurado-Rivera JA, Jaume D, Juan C, Pons J. Comparative Mitogenomics in Hyalella (Amphipoda: Crustacea). Genes. 2021; 12(2):292. https://doi.org/10.3390/genes12020292
Chicago/Turabian StyleZapelloni, Francesco, José A. Jurado-Rivera, Damià Jaume, Carlos Juan, and Joan Pons. 2021. "Comparative Mitogenomics in Hyalella (Amphipoda: Crustacea)" Genes 12, no. 2: 292. https://doi.org/10.3390/genes12020292
APA StyleZapelloni, F., Jurado-Rivera, J. A., Jaume, D., Juan, C., & Pons, J. (2021). Comparative Mitogenomics in Hyalella (Amphipoda: Crustacea). Genes, 12(2), 292. https://doi.org/10.3390/genes12020292