
Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Analysis of RNA Modifications by NGS
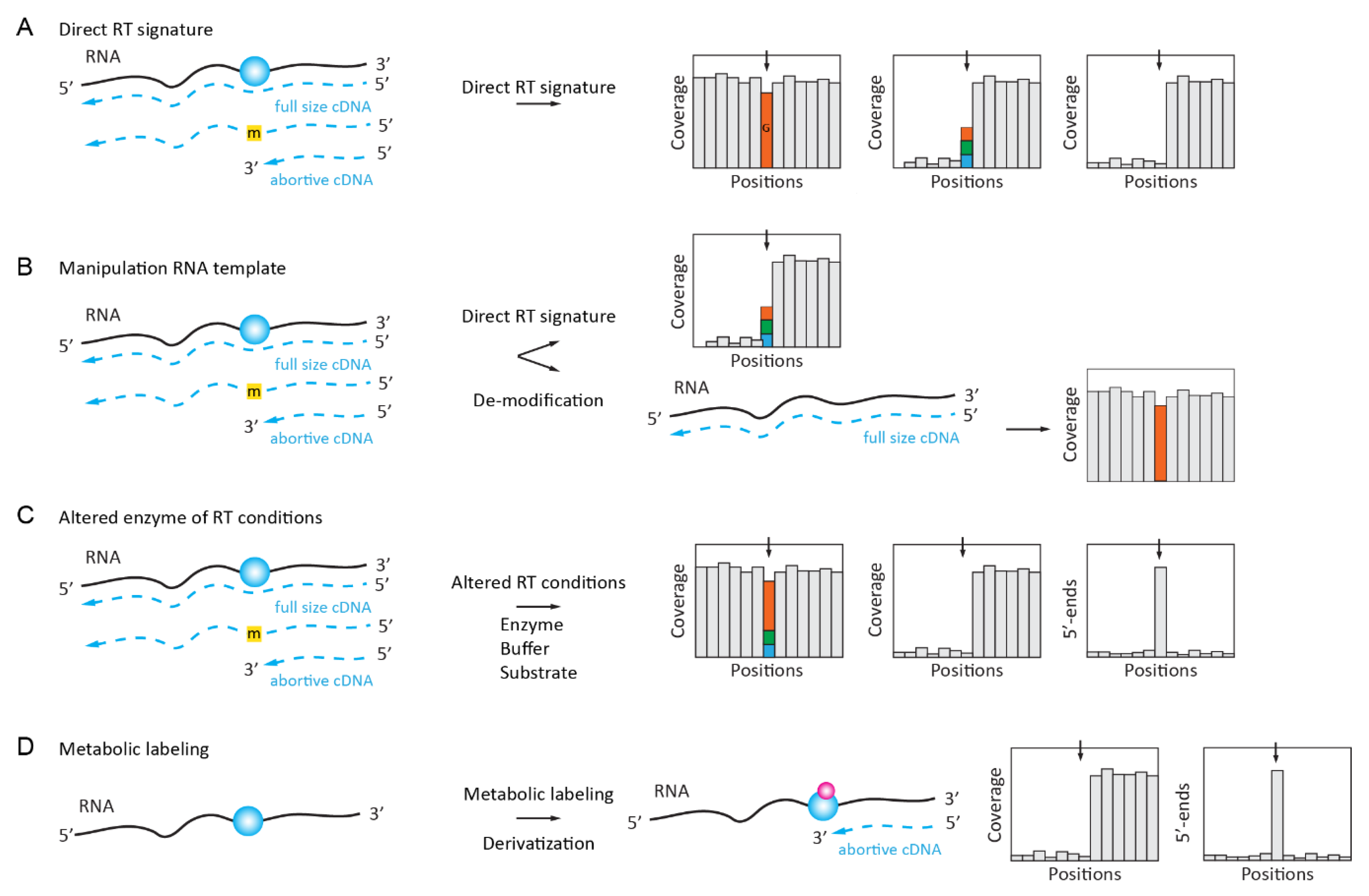
2.1. Naturally Existing RT Signatures of Modified Nucleotides
2.1.1. Inosine “Mutation” RT Signature
2.1.2. Complex RT Signatures for m1A (m3U, m3C, m22G, etc.)
2.2. Enzymatically Enhanced Natural Signatures
2.2.1. Manipulation (De-Modification) of the RNA Template
2.2.2. Manipulation of the Conditions for Enzymatic Reaction or the Nature of dNTP Substrate
2.2.3. Manipulation of the RT Enzyme Properties
2.2.4. Manipulation of RNA Template by in Vivo Metabolic Labeling
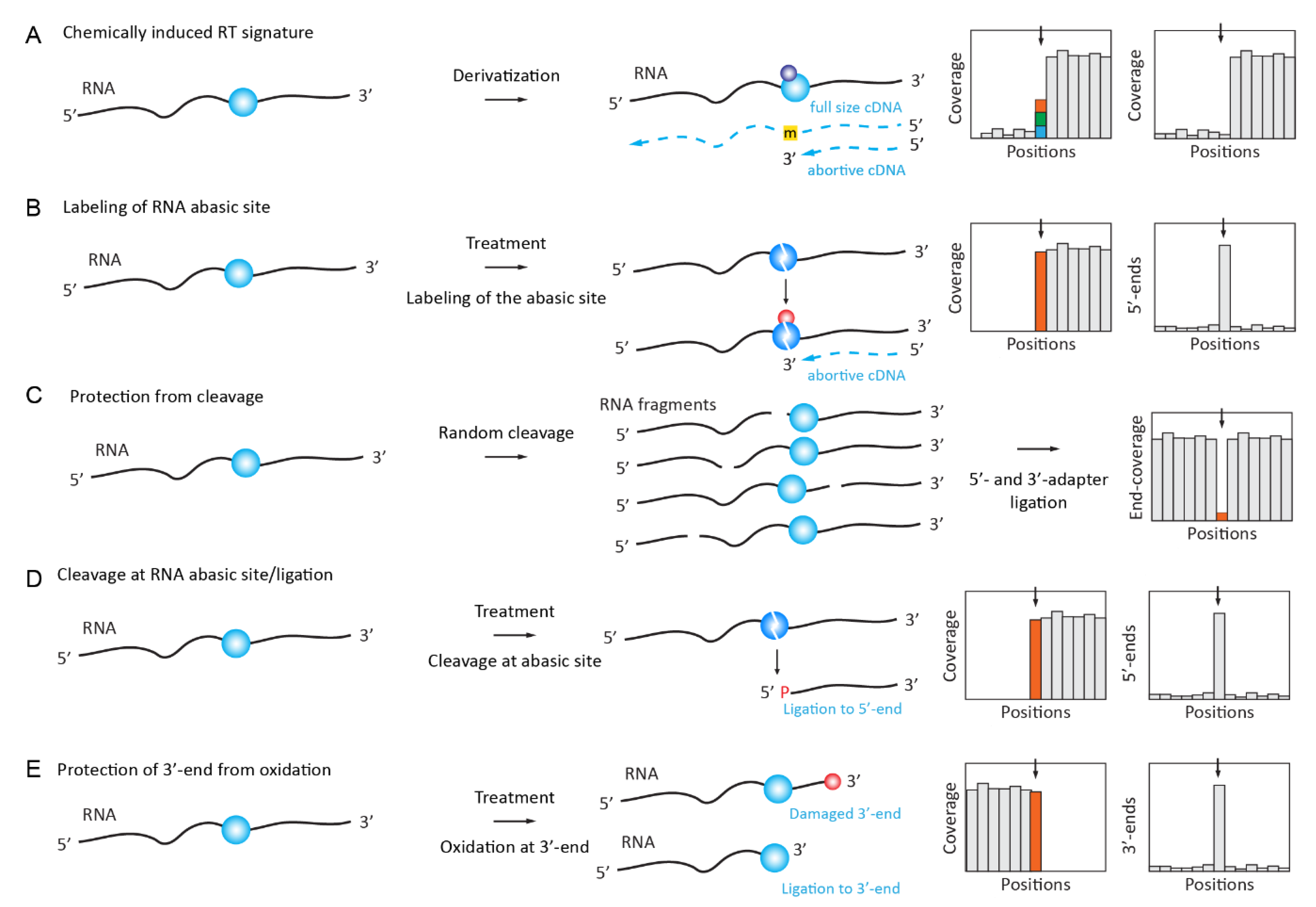
2.3. Chemically Induced RT Signatures or RT Stops
2.3.1. N1-Alkylation of Inosine (I)
2.3.2. S4-Alkylation of 4-Thiouridine (s4U)
2.3.3. Deamination and Oxidation of 5-methylcytosine (m5C)
2.3.4. Derivatization of Pseudouridine (Ψ) by Soluble Carbodiimide
2.3.5. Dimroth Rearrangement of 1-Methyladenosine (m1A) to m6A under Alkaline Conditions
2.3.6. Sodium Borohydride Reduction of 4-Acetylcytidine (ac4C)
2.3.7. Sodium Borohydride Reduction of 7-Methylguanosine (m7G)
2.4. Chemically Induced Cleavage of the Ribose-Phosphate Backbone and Selective Ligation
2.4.1. Detection and Quantification of Nm Residues by RiboMethSeq
2.4.2. Detection of 7-methylguanosine (m7G) by TRAC-Seq and AlkAnilineSeq
2.4.3. Mapping and Quantification of Pseudouridine (Ψ) by HydraPsiSeq
2.4.4. Profiling of m3C Using Hydrazine Cleavage
2.4.5. Detection of Nm RNA Residues by Their Resistance to IO4 Oxidation (RibOxi-Seq/Nm-Seq)
2.4.6. Selective Protection of m6A Methylated Motifs against MazF Cleavage (MAZTER-Seq)
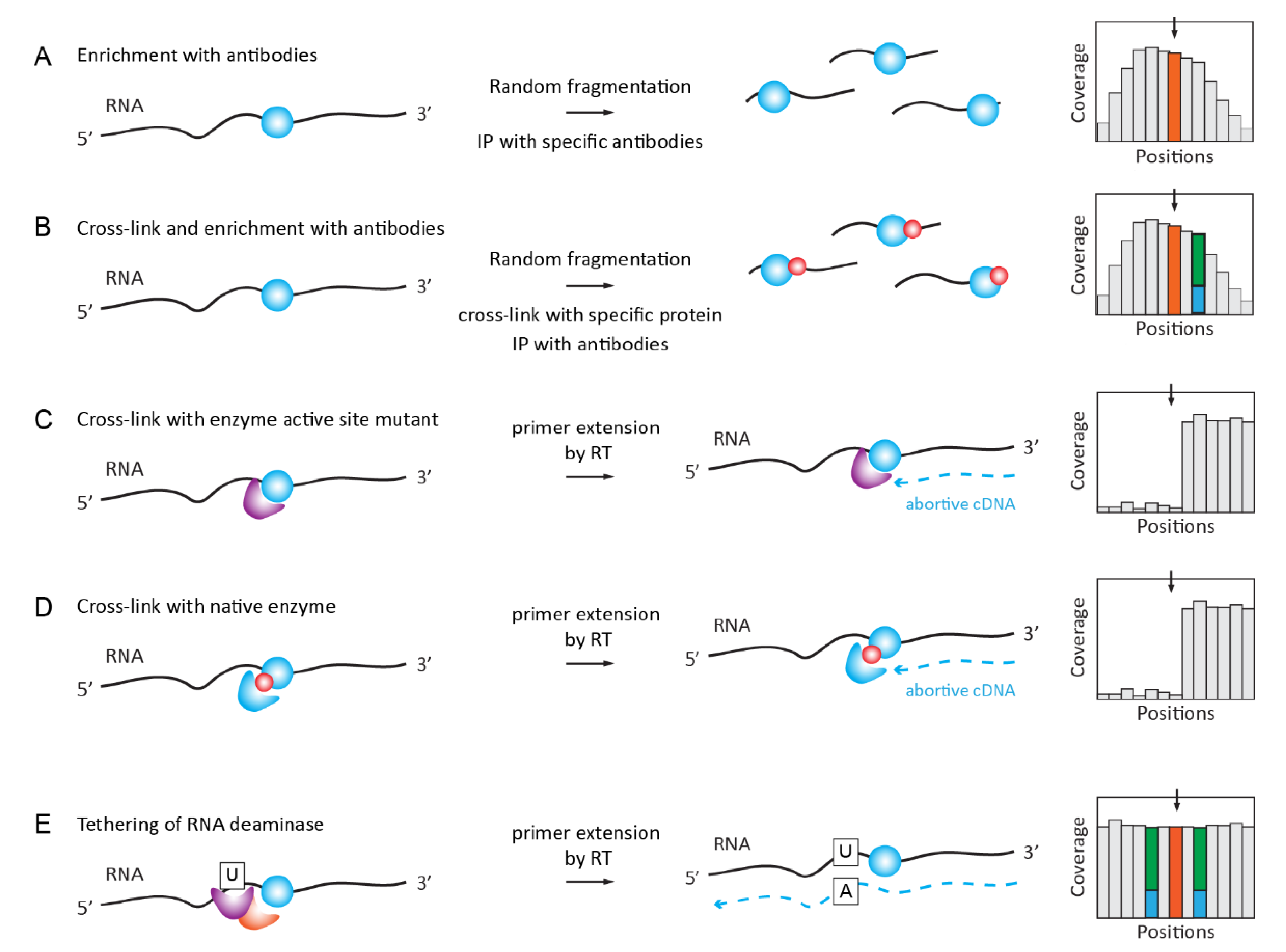
2.5. Antibody-Based Enrichment Methods (MeRIP-Seq, i/miCLIP)
2.5.1. RNA ImmunoPrecipitation (RIP) for m6A, m1A, hm5C, ac4C
2.5.2. iCLIP/PAR-CLIP with Antibodies and Specific Proteins/Enzymes
2.5.3. Covalent Cross-Linking with RNA Target Mediated by RNA Modification Enzyme or Specific Reader Protein
2.6. Data Analysis and Interpretation
3. Analysis of RNA Modifications by NNGS (Single-Molecule Sequencing)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiener, D.; Schwartz, S. The Epitranscriptome beyond m6A. Nat. Rev. Genet. 2020, 1–13. [Google Scholar] [CrossRef]
- Helm, M.; Motorin, Y. Detecting RNA Modifications in the Epitranscriptome: Predict and Validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef]
- Motorin, Y.; Helm, M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes 2019, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Nielsen, H. Sequencing-Based Methods for Detection and Quantitation of Ribose Methylations in RNA. Methods 2019, 156, 5–15. [Google Scholar] [CrossRef]
- Hartstock, K.; Rentmeister, A. MappingN6-Methyladenosine (m6A) in RNA: Established Methods, Remaining Challenges, and Emerging Approaches. Chem. A Eur. J. 2019, 25, 3455–3464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-Y.; Song, J.; Liu, Y.; Song, C.-X.; Yi, C. Mapping the Epigenetic Modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef]
- Linder, B.; Jaffrey, S.R. Discovering and Mapping the Modified Nucleotides That Comprise the Epitranscriptome of mRNA. Cold Spring Harb. Perspect. Biol. 2019, 11, a032201. [Google Scholar] [CrossRef]
- Ouyang, Z.; Ren, C.; Liu, F.; An, G.; Bo, X.; Shu, W. The Landscape of the A-to-I RNA Editome from 462 Human Genomes. Sci. Rep. 2018, 8, 12069. [Google Scholar] [CrossRef] [PubMed]
- Oakes, E.; Vadlamani, P.; Hundley, H.A. Methods for the Detection of Adenosine-to-Inosine Editing Events in Cellular RNA. In Advanced Structural Safety Studies; Springer International Publishing: Berlin, Germany, 2017; Volume 1648, pp. 103–127. [Google Scholar]
- Hauenschild, R.; Tserovski, L.; Schmid, K.; Thuring, K.L.; Winz, M.-L.; Sharma, S.; Entian, K.-D.; Wacheul, L.; Lafontaine, D.L.J.; Anderson, J.; et al. The reverse Transcription Signature ofN-1-Methyladenosine in RNA-Seq is Sequence Dependent. Nucleic Acids Res. 2015, 43, 9950–9964. [Google Scholar] [CrossRef] [PubMed]
- Tserovski, L.; Marchand, V.; Hauenschild, R.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. High-Throughput Sequencing for 1-Methyladenosine (m1a) Mapping in RNA. Methods 2016, 107, 110–121. [Google Scholar] [CrossRef]
- Ryvkin, P.; Leung, Y.Y.; Silverman, I.M.; Childress, M.; Valladares, O.; Dragomir, I.; Gregory, B.D.; Wang, L.-S. HAMR: High-Throughput Annotation of Modified Ribonucleotides. RNA 2013, 19, 1684–1692. [Google Scholar] [CrossRef]
- Kuksa, P.P.; Leung, Y.Y.; Vandivier, L.E.; Anderson, Z.; Gregory, B.D.; Wang, L.-S. In Silico Identification of RNA Modifications from High-Throughput Sequencing Data Using HAMR. Methods Mol. Biol. 2017, 1562, 211–229. [Google Scholar] [CrossRef]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A Landscape on Cytosolic and Mitochondrial mRNA at Single-Base Resolution. Nat. Cell Biol. 2017, 551, 251–255. [Google Scholar] [CrossRef]
- Kietrys, A.M.; Velema, W.A.; Kool, E.T. Fingerprints of Modified RNA Bases from Deep Sequencing Profiles. J. Am. Chem. Soc. 2017, 139, 17074–17081. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Toffano-Nioche, C.; Lorieux, F.; Gautheret, D.; Lehmann, J. Accurate Characterization of Escherichia Coli Trna Modifications with a Simple Method of Deep-Sequencing Library Preparation. RNA Biol. 2021, 18, 33–46. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-Seq: AlkB-Facilitated RNA Methylation Sequencing Reveals a Complex Landscape of Modified tRNA Fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and Quantitative High-Throughput rRNA Sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Zheng, G.; Schwartz, M.H.; Clark, W.C.; Pan, T. Selective Enzymatic Demethylation of N 2, N 2-Dimethylguanosine in RNA and Its Application in High-Throughput tRNA Sequencing. Angew. Chem. Int. Ed. 2017, 56, 5017–5020. [Google Scholar] [CrossRef]
- Schwartz, M.H.; Wang, H.; Pan, J.N.; Clark, W.C.; Cui, S.; Eckwahl, M.J.; Pan, D.W.; Parisien, M.; Owens, S.M.; Cheng, B.L.; et al. Microbiome Characterization by High-Throughput Transfer RNA Sequencing and Modification Analysis. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Dong, S.; Yu, Q.; Jia, G. Antibody-Free Enzyme-Assisted Chemical Approach for Detection of N6-Methyladenosine. Nat. Chem. Biol. 2020, 16, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Sendinc, E.; Valle-Garcia, D.; Dhall, A.; Chen, H.; Henriques, T.; Navarrete-Perea, J.; Sheng, W.; Gygi, S.P.; Adelman, K.; Shi, Y. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol. Cell 2019, 75, 620–630.e9. [Google Scholar] [CrossRef]
- Behm-Ansmant, I.; Helm, M.; Motorin, Y. Use of Specific Chemical Reagents for Detection of Modified Nucleotides in RNA. J. Nucleic Acids 2011, 2011, 1–17. [Google Scholar] [CrossRef]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of Modified Residues in RNAs by Reverse Transcription-Based Methods. Methods Enzymol. 2007, 425, 21–53. [Google Scholar] [CrossRef]
- Maden, B.H. Mapping 2′-O-Methyl Groups in Ribosomal RNA. Methods 2001, 25, 374–382. [Google Scholar] [CrossRef]
- Incarnato, D.; Anselmi, F.; Morandi, E.; Neri, F.; Maldotti, M.; Rapelli, S.; Parlato, C.; Basile, G.; Oliviero, S. High-Throughput Single-Base Resolution Mapping of RNA 2’-O-Methylated Residues. Nucleic Acids Res. 2017, 45, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Kristen, M.; Plehn, J.; Marchand, V.; Friedland, K.; Motorin, Y.; Helm, M.; Werner, S. Manganese Ions Individually Alter the Reverse Transcription Signature of Modified Ribonucleosides. Genes 2020, 11, 950. [Google Scholar] [CrossRef]
- Hong, T.; Yuan, Y.; Chen, Z.; Xi, K.; Wang, T.; Xie, Y.; He, Z.; Su, H.; Zhou, Y.; Tan, Z.-J.; et al. Precise Antibody-Independent m6A Identification via 4SedTTP-Involved and FTO-Assisted Strategy at Single-Nucleotide Resolution. J. Am. Chem. Soc. 2018, 140, 5886–5889. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, E.M.; Ehrenschwender, T.; Batista, P.J.; Chang, H.Y.; Kool, E.T. Identification of a Selective Polymerase Enables Detection of N6-Methyladenosine in RNA. J. Am. Chem. Soc. 2013, 135, 19079–19082. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Y.; Tang, Q.; Wei, L.; Zhang, X.; Jia, G. An Elongation-and Ligation-Based qPCR Amplification Method for the Radiolabeling-Free Detection of Locus-Specific N 6 -Methyladenosine Modification. Angew. Chem. Int. Ed. 2018, 57, 15995–16000. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Z.; Sepich-Poore, C.; Zhang, L.; Xiao, Y.; He, C. LEAD-m 6 A-seq for Locus-Specific Detection of N 6-Methyladenosine and Quantification of Differential Methylation. Angew. Chem. Int. Ed. 2021, 60, 873–880. [Google Scholar] [CrossRef]
- Dong, Z.-W.; Shao, P.; Diao, L.-T.; Zhou, H.; Yu, C.-H.; Qu, L.-H. RTL-P: A Sensitive Approach for Detecting Sites of 2′-O-Methylation in RNA Molecules. Nucleic Acids Res. 2012, 40, e157. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, J.; Marx, A. Direct and Site-Specific Quantification of RNA 2′-O-Methylation by PCR with an Engineered DNA Polymerase. Nucleic Acids Res. 2016, 44, 3495–3502. [Google Scholar] [CrossRef]
- Aschenbrenner, J.; Werner, S.; Marchand, V.; Adam, M.; Motorin, Y.; Helm, M.; Marx, A. Engineering of a DNA Polymerase for Direct m6A Sequencing. Angew. Chem. Int. Ed. 2018, 57, 417–421. [Google Scholar] [CrossRef]
- Zhou, H.; Rauch, S.; Dai, Q.; Cui, X.; Zhang, Z.; Nachtergaele, S.; Sepich, C.; He, C.; Dickinson, B.C. Evolution of a Reverse Transcriptase to Map N1-Methyladenosine in Human Messenger RNA. Nat. Methods 2019, 16, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Burhenne, J.; Teimer, R.; Koynov, K.; Willnow, S.; Weinhold, E.; Helm, M. Expanding the Chemical Scope of RNA: Methyltransferases to Site-Specific Alkynylation of RNA for Click Labeling. Nucleic Acids Res. 2010, 39, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Hartstock, K.; Nilges, B.S.; Ovcharenko, A.; Cornelissen, N.V.; Puellen, N.; Lawrence-Dörner, A.-M.; Leidel, S.A.; Rentmeister, A. Enzymatic or In Vivo Installation of Propargyl Groups in Combination with Click Chemistry for the Enrichment and Detection of Methyltransferase Target Sites in RNA. Angew. Chem. Int. Ed. 2018, 57, 6342–6346. [Google Scholar] [CrossRef]
- Shu, X.; Cao, J.; Cheng, M.; Xiang, S.; Gao, M.; Li, T.; Ying, X.; Wang, F.; Yue, Y.; Lu, Z.; et al. A Metabolic Labeling Method Detects m6A Transcriptome-Wide at Single Base Resolution. Nat. Chem. Biol. 2020, 16, 887–895. [Google Scholar] [CrossRef]
- Holstein, J.M.; Rentmeister, A. Current Covalent Modification Methods for Detecting RNA in Fixed and Living Cells. Methods 2016, 98, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ovcharenko, A.; Rentmeister, A. Emerging Approaches for Detection of Methylation Sites in RNA. Open Biol. 2018, 8, 8. [Google Scholar] [CrossRef]
- Muthmann, N.; Hartstock, K.; Rentmeister, A. Chemo-Enzymatic Treatment of RNA to Facilitate Analyses. Wiley Interdiscip. Rev. RNA 2020, 11, e1561. [Google Scholar] [CrossRef]
- Suzuki, T.; Ueda, H.; Okada, S.; Sakurai, M. Transcriptome-Wide Identification of Adenosine-to-Inosine Editing Using the ICE-Seq Method. Nat. Protoc. 2015, 10, 715–732. [Google Scholar] [CrossRef]
- Knutson, S.D.; Ayele, T.M.; Heemstra, J.M. Chemical Labeling and Affinity Capture of Inosine-Containing RNAs Using Acrylamidofluorescein. Bioconjugate Chem. 2018, 29, 2899–2903. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-Wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Herzog, V.A.; Reichholf, B.; Neumann, T.; Rescheneder, P.; Bhat, P.; Burkard, T.R.; Wlotzka, W.; Von Haeseler, A.; Zuber, J.; Ameres, S.L. Thiol-Linked Alkylation of RNA to Assess Expression Dynamics. Nat. Methods 2017, 14, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Riml, C.; Amort, T.; Rieder, D.; Gasser, C.; Lusser, A.; Micura, R. Osmium-Mediated Transformation of 4-Thiouridine to Cytidine as Key to Study RNA Dynamics by Sequencing. Angew. Chem. Int. Ed. 2017, 56, 13479–13483. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA Cytosine Methylation Analysis by Bisulfite Sequencing. Nucleic Acids Res. 2008, 37, e12. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M. RNA 5-Methylcytosine Analysis by Bisulfite Sequencing. Methods Enzymol. 2015, 560, 297–329. [Google Scholar] [CrossRef]
- Bourgeois, G.; Ney, M.; Gaspar, I.; Aigueperse, C.; Schaefer, M.; Kellner, S.; Helm, M.; Motorin, Y. Eukaryotic rRNA Modification by Yeast 5-Methylcytosine-Methyltransferases and Human Proliferation-Associated Antigen p120. PLoS ONE 2015, 10, e0133321. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Hartmann, M.; Schuster, I.; Bender, S.; Thüring, K.L.; Helm, M.; Katze, J.R.; Nellen, W.; Lyko, F.; Ehrenhofer-Murray, A.E. Dynamic Modulation of Dnmt2-Dependent tRNA Methylation by the Micronutrient Queuine. Nucleic Acids Res. 2015, 43, 10952–10962. [Google Scholar] [CrossRef]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA Cytosine Methylation by Dnmt2 and NSun2 Promotes tRNA Stability and Protein Synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Amort, T.; Rieder, D.; Wille, A.; Khokhlova-Cubberley, D.; Riml, C.; Trixl, L.; Jia, X.-Y.; Micura, R.; Lusser, A. Distinct 5-Methylcytosine Profiles in Poly(A) RNA from Mouse Embryonic Stem Cells and Brain. Genome Biol. 2017, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- David, R.; Burgess, A.; Parker, B.; Li, J.; Pulsford, K.; Sibbritt, T.; Preiss, T.; Searle, I.R. Transcriptome-Wide Mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and Noncoding RNAs. Plant Cell 2017, 29, 445–460. [Google Scholar] [CrossRef]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-Wide Mapping of 5-Methylcytidine RNA Modifications in Bacteria, Archaea, and Yeast Reveals m5C within Archaeal mRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef]
- Wei, Z.; Panneerdoss, S.; Timilsina, S.; Zhu, J.; Mohammad, T.A.; Lu, Z.-L.; De Magalhães, J.P.; Chen, Y.; Rong, R.; Huang, Y.; et al. Topological Characterization of Human and Mouse m5C Epitranscriptome Revealed by Bisulfite Sequencing. Int. J. Genom. 2018, 2018, 1–19. [Google Scholar] [CrossRef]
- Legrand, C.; Tuorto, F.; Hartmann, M.; Liebers, R.; Jacob, D.; Helm, M.; Lyko, F. Statistically Robust Methylation Calling for Whole-Transcriptome Bisulfite Sequencing Reveals Distinct Methylation Patterns for Mouse RNAs. Genome Res. 2017, 27, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-S.; Ma, H.-L.; Yang, Y.; Lai, W.-Y.; Sun, B.-F.; Yang, Y.-G. 5-Methylcytosine Analysis by RNA-BisSeq. In Advanced Structural Safety Studies; Springer International Publishing: Berlin, Germany, 2018; Volume 1870, pp. 237–248. [Google Scholar]
- Khoddami, V.; Yerra, A.; Mosbruger, T.L.; Fleming, A.M.; Burrows, C.J.; Cairns, B.R. Transcriptome-Wide Profiling of Multiple RNA Modifications Simultaneously at Single-Base Resolution. Proc. Natl. Acad. Sci. USA 2019, 116, 6784–6789. [Google Scholar] [CrossRef]
- Song, C.-X.; Szulwach, K.E.; Dai, Q.; Fu, Y.; Mao, S.-Q.; Lin, L.; Street, C.; Li, Y.; Poidevin, M.; Wu, H.; et al. Genome-Wide Profiling of 5-Formylcytosine Reveals Its Roles in Epigenetic Priming. Cell 2013, 153, 678–691. [Google Scholar] [CrossRef]
- Booth, M.J.; Marsico, G.; Bachman, M.; Beraldi, D.; Balasubramanian, S. Quantitative Sequencing of 5-Formylcytosine in DNA at Single-Base Resolution. Nat. Chem. 2014, 6, 435–440. [Google Scholar] [CrossRef]
- Van Haute, L.; Dietmann, S.; Kremer, L.; Hussain, S.; Pearce, S.F.; Powell, C.A.; Rorbach, J.; Lantaff, R.; Blanco, S.; Sauer, S.; et al. Deficient Methylation and Formylation of mt-tRNAMet Wobble Cytosine in a Patient Carrying Mutations in NSUN3. Nat. Commun. 2016, 7, 12039. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Bi, Y.; Siejka-Zielinska, P.; Zhou, Y.-L.; Zhang, X.-X.; Song, C.-X. Bisulfite-Free and Base-Resolution Analysis of 5-Methylcytidine and 5-Hydroxymethylcytidine in RNA with Peroxotungstate. Chem. Commun. 2019, 55, 2328–2331. [Google Scholar] [CrossRef]
- Zeng, H.; He, B.; Xia, B.; Bai, D.; Lu, X.; Cai, J.; Chen, L.; Zhou, A.; Zhu, C.; Meng, H.; et al. Bisulfite-Free, Nanoscale Analysis of 5-Hydroxymethylcytosine at Single Base Resolution. J. Am. Chem. Soc. 2018, 140, 13190–13194. [Google Scholar] [CrossRef]
- Werner, S.; Galliot, A.; Pichot, F.; Kemmer, T.; Marchand, V.; Sednev, M.V.; Lence, T.; Roignant, J.-Y.; König, J.; Höbartner, C.; et al. NOseq: Amplicon Sequencing Evaluation Method for RNA m6A Sites after Chemical Deamination. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Bakin, A.; Ofengand, J. Four Newly Located Pseudouridylate Residues in Escherichia Coli 23S Ribosomal RNA Are All at the Peptidyltransferase Center: Analysis by the Application of a New Sequencing Technique. Biochemical 1993, 32, 9754–9762. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine Profiling Reveals Regulated mRNA Pseudouridylation in Yeast and Human Cells. Nat. Cell Biol. 2014, 515, 143–146. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Transcriptome-Wide Identification of Pseudouridine Modifications Using Pseudo-Seq. Curr. Protoc. Mol. Biol. 2015, 112, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Pseudo-Seq. Methods Enzymol. 2015, 560, 219–245. [Google Scholar] [CrossRef]
- Lovejoy, A.F.; Riordan, D.P.; Brown, P.O. Transcriptome-Wide Mapping of Pseudouridines: Pseudouridine Synthases Modify Specific mRNAs in S. cerevisiae. PLOS ONE 2014, 9, e110799. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, M.A.; Lovejoy, A.F.; Cygan, A.M.; Boothroyd, J.C. mRNA Pseudouridylation Affects RNA Metabolism in the Parasite Toxoplasma gondii. RNA 2017, 23, 1834–1849. [Google Scholar] [CrossRef]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-Wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Rajan, K.S.; Doniger, T.; Cohen-Chalamish, S.; Chen, D.; Semo, O.; Aryal, S.; Saar, E.G.; Chikne, V.; Gerber, D.; Unger, R.; et al. Pseudouridines on Trypanosoma Brucei Spliceosomal Small Nuclear RNAs and Their Implication for RNA and Protein Interac-Tions. Nucleic Acids Res. 2019, 47, 7633–7647. [Google Scholar] [CrossRef]
- Li, X.; Zhu, P.; Ma, S.; Song, J.; Bai, J.; Sun, F.; Yi, C. Chemical Pulldown Reveals Dynamic Pseudouridylation of the Mammalian Transcriptome. Nat. Chem. Biol. 2015, 11, 592–597. [Google Scholar] [CrossRef]
- Li, X.; Ma, S.; Yi, C. Pseudouridine Chemical Labeling and Profiling. Methods Enzymol. 2015, 560, 247–272. [Google Scholar] [CrossRef]
- Zaringhalam, M.; Papavasiliou, F.N. Pseudouridylation Meets Next-Generation Sequencing. Methods 2016, 107, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Nir, R.; Farouq, D.; Slutskin, I.V.; Schwartz, S. TRUB1 Is the Predominant Pseudouridine Synthase Acting on Mammalian mRNA via a Predictable and Conserved Code. Genome Res. 2017, 27, 393–406. [Google Scholar] [CrossRef]
- Zhou, K.I.; Clark, W.C.; Pan, D.W.; Eckwahl, M.J.; Dai, Q.; Pan, T. Pseudouridines Have Context-Dependent Mutation and Stop Rates in High-Throughput Sequencing. RNA Biol. 2018, 15, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The Dynamic N1-Methyladenosine Methylome in Eukaryotic Messenger RNA. Nat. Cell Biol. 2016, 530, 441–446. [Google Scholar] [CrossRef]
- Thomas, J.M.; Briney, C.A.; Nance, K.D.; Lopez, J.E.; Thorpe, A.L.; Fox, S.D.; Bortolin-Cavaille, M.-L.; Sas-Chen, A.; Arango, D.; Oberdoerffer, S.; et al. A Chemical Signature for Cytidine Acetylation in RNA. J. Am. Chem. Soc. 2018, 140, 12667–12670. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Bryson, K.M.; Meier, J.L. Nucleotide Resolution Sequencing of N4-Acetylcytidine in RNA. Methods Enzymol. 2019, 621, 31–51. [Google Scholar] [CrossRef] [PubMed]
- Sas-Chen, A.; Thomas, J.M.; Matzov, D.; Taoka, M.; Nance, K.D.; Nir, R.; Bryson, K.M.; Shachar, R.; Liman, G.L.S.; Burkhart, B.W.; et al. Dynamic RNA Acetylation Revealed by Quantitative Cross-Evolutionary Mapping. Nat. Cell Biol. 2020, 583, 638–643. [Google Scholar] [CrossRef]
- Wintermeyer, W.; Zachau, H.G. Tertiary Structure Interactions of 7-Methylguanosine in Yeast TRNA Phe as Studied by Borohydride Reduction. FEBS Lett. 1975, 58, 306–309. [Google Scholar] [CrossRef]
- Peattie, D.A. Direct Chemical Method for Sequencing RNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Zueva, V.S.; Mankin, A.S.; Bogdanov, A.A.; Baratova, L.A. Specific Fragmentation of tRNA and rRNA at a 7-Methylguanine Residue in the Presence of Methylated Carrier RNA. JBIC J. Biol. Inorg. Chem. 1985, 146, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Enroth, C.; Poulsen, L.D.; Iversen, S.; Kirpekar, F.; Albrechtsen, A.; Vinther, J. Detection of Internal N7-Methylguanosine (m7G) RNA Modifications by Mutational Profiling Sequencing. Nucleic Acids Res. 2019, 47, e126. [Google Scholar] [CrossRef] [PubMed]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C.; et al. METTL1 Promotes Let-7 MicroRNA Processing via m7G Methylation. Mol. Cell 2019, 74, 1278–1290.e9. [Google Scholar] [CrossRef] [PubMed]
- Vinther, J. No Evidence for N7-Methylation of Guanosine (m7G) in Human Let-7e. Mol. Cell 2020, 79, 199–200. [Google Scholar] [CrossRef]
- Kouzarides, T.; Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Andrews, B. Further Evidence Supporting N7-Methylation of Guanosine (m7G) in Human MicroRNAs. Mol. Cell 2020, 79, 201–202. [Google Scholar] [CrossRef]
- Birkedal, U.; Christensen-Dalsgaard, M.; Krogh, N.; Sabarinathan, R.; Gorodkin, J.; Nielsen, H. Profiling of Ribose Methylations in RNA by High-Throughput Sequencing. Angew. Chem. Int. Ed. 2014, 54, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Gumienny, R.; Jedlinski, D.J.; Schmidt, A.; Gypas, F.; Martin, G.; Vina-Vilaseca, A.; Zavolan, M. High-Throughput Identification of C/D box snoRNA Targets with CLIP and RiboMeth-seq. Nucleic Acids Res. 2016, 45, 2341–2353. [Google Scholar] [CrossRef]
- Marchand, V.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. Illumina-Based RiboMethSeq Approach for Mapping of 2′-O-Me Residues in RNA. Nucleic Acids Res. 2016, 44, e135. [Google Scholar] [CrossRef]
- Marchand, V.; Ayadi, L.; El Hajj, A.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. High-Throughput Mapping of 2′-O-Me Residues in RNA Using Next-Generation Sequencing (Illumina RiboMethSeq Protocol). Breast Cancer 2017, 1562, 171–187. [Google Scholar] [CrossRef]
- Ringeard, M.; Marchand, V.; Decroly, E.; Motorin, Y.; Bennasser, Y. FTSJ3 is an RNA 2′-O-Methyltransferase Recruited by HIV to Avoid Innate Immune Sensing. Nat. Cell Biol. 2019, 565, 500–504. [Google Scholar] [CrossRef]
- Erales, J.; Marchand, V.; Panthu, B.; Gillot, S.; Belin, S.; Ghayad, S.E.; Garcia, M.; Laforêts, F.; Marcel, V.; Baudin-Baillieu, A.; et al. Evidence for rRNA 2′-O-Methylation Plasticity: Control of Intrinsic Translational Capabilities of Human Ribosomes. Proc. Natl. Acad. Sci. USA 2017, 114, 12934–12939. [Google Scholar] [CrossRef]
- Hebras, J.; Krogh, N.; Marty, V.; Nielsen, H.; Cavaillé, J. Developmental Changes of rRNA Ribose Methylations in the Mouse. RNA Biol. 2020, 17, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Freund, I.; Buhl, D.K.; Boutin, S.; Kotter, A.; Pichot, F.; Marchand, V.; Vierbuchen, T.; Heine, H.; Motorin, Y.; Helm, M.; et al. 2′-O-Methylation within Prokaryotic and Eukaryotic tRNA Inhibits Innate Immune Activation by Endosomal Toll-like Receptors but Does Not Affect Recognition of Whole Organisms. RNA 2019, 25, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Rajan, K.S.; Zhu, Y.; Adler, K.; Doniger, T.; Cohen-Chalamish, S.; Srivastava, A.; Shalev-Benami, M.; Matzov, D.; Unger, R.; Tschudi, C.; et al. The Large Repertoire of 2’-O-Methylation Guided by C/D snoRNAs on Trypanosoma Brucei rRNA. RNA Biol. 2020, 17, 1018–1039. [Google Scholar] [CrossRef]
- Ramachandran, S.; Krogh, N.; Jørgensen, T.E.; Johansen, S.D.; Nielsen, H.; Babiak, I. The Shift from Early to Late Types of Ribosomes in Zebrafish Development Involves Changes at a Subset of rRNA 2′-O-Me Sites. RNA 2020, 26, 1919–1934. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.T.; Dimitrova, D.G.; Da Silva, B.; Marchand, V.; Jacquier, C.; Achour, C.; Brazane, M.; Goyenvalle, C.; Bourguignon-Igel, V.; Shehzada, S.; et al. tRNA 2′-O-Methylation by a Duo of TRM7/FTSJ1 Proteins Modulates Small RNA Silencing in Drosophila. Nucleic Acids Res. 2020, 48, 2050–2072. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Liu, Q.; Jiang, Y.-Z.; Gregory, R.I. Nucleotide Resolution Profiling of m7G tRNA Modification by TRAC-Seq. Nat. Protoc. 2019, 14, 3220–3242. [Google Scholar] [CrossRef]
- Lin, S.; Liu, Q.; Lelyveld, V.S.; Choe, J.; Szostak, J.W.; Gregory, R.I. Mettl1/Wdr4-Mediated m7G tRNA Methylome Is Required for Normal mRNA Translation and Embryonic Stem Cell Self-Renewal and Differentiation. Mol. Cell 2018, 71, 244–255.e5. [Google Scholar] [CrossRef] [PubMed]
- Marchand, V.; Ayadi, L.; Ernst, F.G.M.; Hertler, J.; Bourguignon-Igel, V.; Galvanin, A.; Kotter, A.; Helm, M.; Lafontaine, D.L.J.; Motorin, Y. AlkAniline-Seq: Profiling of m 7 G and m 3 C RNA Modifications at Single Nucleotide Resolution. Angew. Chem. Int. Ed. 2018, 57, 16785–16790. [Google Scholar] [CrossRef]
- Marchand, V.; Pichot, F.; Neybecker, P.; Ayadi, L.; Bourguignon-Igel, V.; Wacheul, L.; Lafontaine, D.L.J.; Pinzano, A.; Helm, M.; Motorin, Y. HydraPsiSeq: A Method for Systematic and Quantitative Mapping of Pseudouridines in RNA. Nucleic Acids Res. 2020, 48, e110. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Liu, Q.; Sendinc, E.; Shi, Y.; I Gregory, R. Nucleotide Resolution Profiling of m3C RNA Modification by HAC-seq. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq Maps 2′-O-Methylation Sites in Human mRNA with Base Precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pirnie, S.P.; Carmichael, G.G. High-Throughput and Site-Specific Identification of 2′-O-Methylation Sites Using Ribose Oxidation Sequencing (RibOxi-seq). RNA 2017, 23, 1303–1314. [Google Scholar] [CrossRef]
- Pandey, R.R.; Pillai, R.S. Counting the Cuts: MAZTER-Seq Quantifies m6A Levels Using a Methylation-Sensitive Ribonuclease. Cell 2019, 178, 515–517. [Google Scholar] [CrossRef]
- Milstone, D.S.; Vold, B.S.; Glitz, D.G.; Shutt, N. Antibodies to N6-(Δ2-isopentenyl) Adenosine and Its Nucleotide: Interaction with Purified tRNAs and with Bases, Nucleosides and Nucleotides of the Isopen-Tenyladenosine Family. Nucleic Acids Res. 1978, 5, 3439–3455. [Google Scholar] [CrossRef]
- Vold, B.S.; Nolen, H.W. A Unique Method Utilizing Antinucleotide Antibodies for Evaluating Changes in the Levels of Modified Nucleosides of tRNAs from Crude Extracts of Whole Cells. Nucleic Acids Res. 1979, 7, 971–980. [Google Scholar] [CrossRef][Green Version]
- Woodsworth, M.L.; Latimer, L.J.; Janzer, J.J.; McLennan, B.D.; Lee, J.S. Characierization of Monoclonal Antibodies Specific for Isopentenyl Adenosine Derivatives Occurring in Transfer RNA. Biochem. Biophys. Res. Commun. 1983, 114, 791–796. [Google Scholar] [CrossRef]
- Feederle, R.; Schepers, A. Antibodies Specific for Nucleic Acid Modifications. RNA Biol. 2017, 14, 1089–1098. [Google Scholar] [CrossRef]
- Matsuzawa, S.; Wakata, Y.; Ebi, F.; Isobe, M.; Kurosawa, N. Development and Validation of Monoclonal Antibodies against N6-Methyladenosine for the Detection of RNA Modifications. PLoS ONE 2019, 14, e0223197. [Google Scholar] [CrossRef]
- Mishima, E.; Jinno, D.; Akiyama, Y.; Itoh, K.; Nankumo, S.; Shima, H.; Kikuchi, K.; Takeuchi, Y.; Elkordy, A.; Suzuki, T.; et al. Immuno-Northern Blotting: Detection of RNA Modifications by Using Antibodies against Modified Nucleosides. PLoS ONE 2015, 10, e0143756. [Google Scholar] [CrossRef]
- Slama, K.; Galliot, A.; Weichmann, F.; Hertler, J.; Feederle, R.; Meister, G.; Helm, M. Determination of Enrichment Factors for Modified RNA in MeRIP Experiments. Methods 2019, 156, 102–109. [Google Scholar] [CrossRef]
- Grozhik, A.V.; Olarerin-George, A.O.; Sindelar, M.; Li, X.; Gross, S.S.; Jaffrey, S.R. Antibody Cross-Reactivity Accounts for Widespread Appearance of m1A in 5’UTRs. Nat. Commun. 2019, 10, 5126. [Google Scholar] [CrossRef]
- McIntyre, A.B.R.; Gokhale, N.S.; Cerchietti, L.; Jaffrey, S.R.; Horner, S.M.; Mason, C.E. Limits in the Detection of m6A Changes Using MeRIP/m6A-seq. Sci. Rep. 2020, 10, 6590. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Lyko, F.; Motorin, Y. Limited Antibody Specificity Compromises Epitranscriptomic Analyses. Nat. Commun. 2019, 10, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Divon, S.M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the Human and Mouse m6A RNA Methylomes revealed by m6A-seq. Nat. Cell Biol. 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Salmon-Divon, M.; Amariglio, N.; Rechavi, G. Transcriptome-Wide Mapping of N6-Methyladenosine by m6A-Seq Based on Immunocapturing and Massively Parallel Sequencing. Nat. Protoc. 2013, 8, 176–189. [Google Scholar] [CrossRef]
- Vandivier, L.E.; Gregory, B.D. Reading the Epitranscriptome. Chick Chorioallantoic Membr. Model Precis. Cancer Ther. 2017, 41, 269–298. [Google Scholar] [CrossRef]
- Delatte, B.; Wang, F.; Ngoc, L.V.; Collignon, E.; Bonvin, E.; Deplus, R.; Calonne, E.; Hassabi, B.; Putmans, P.; Awe, S.; et al. Transcriptome-Wide Distribution and Function of RNA Hydroxymethylcytosine. Science 2016, 351, 282–285. [Google Scholar] [CrossRef]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 2018, 175, 1872–1886.e24. [Google Scholar] [CrossRef]
- Malbec, L.; Zhang, T.; Chen, Y.-S.; Sun, B.-F.; Shi, B.-Y.; Zhao, Y.-L.; Yang, Y.; Yang, Y.-G. Dynamic Methylome of Internal mRNA N7-Methylguanosine and Its Regulatory Role in Translation. Cell Res. 2019, 29, 927–941. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-Nucleotide-resolution Mapping of m6A and m6Am throughout the Transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Hawley, B.R.; Jaffrey, S.R. Transcriptome-Wide Mapping of m6A and m6Am at Single-Nucleotide Resolution Using miCLIP. Curr. Protoc. Mol. Biol. 2019, 126, e88. [Google Scholar] [CrossRef] [PubMed]
- Grozhik, A.V.; Linder, B.; Olarerin-George, A.O.; Jaffrey, S.R. Mapping m6A at Individual-Nucleotide Resolution Using Crosslinking and Immunoprecipitation (miCLIP). Methods Mol. Biol. 2017, 1562, 55–78. [Google Scholar] [CrossRef]
- Boulias, K.; Toczydłowska-Socha, D.; Hawley, B.R.; Liberman, N.; Takashima, K.; Zaccara, S.; Guez, T.; Vasseur, J.-J.; Debart, F.; Aravind, L.; et al. Identification of the m6Am Methyltransferase PCIF1 Reveals the Location and Functions of m6Am in the Transcriptome. Mol. Cell 2019, 75, 631–643.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-Methyladenosine-Dependent RNA Structural Switches Regulate RNA–Protein Interactions. Nat. Cell Biol. 2015, 518, 560–564. [Google Scholar] [CrossRef]
- King, M.Y.; Redman, K.L. RNA Methyltransferases Utilize Two Cysteine Residues in the Formation of 5-Methylcytosine. Biochemical 2002, 41, 11218–11225. [Google Scholar] [CrossRef]
- Redman, K.L. Assembly of Protein−RNA Complexes Using Natural RNA and Mutant Forms of an RNA Cytosine Methyltransferase. Biomacromolecules 2006, 7, 3321–3326. [Google Scholar] [CrossRef]
- Moon, H.J.; Redman, K.L. Trm4 and Nsun2 RNA:m5C Methyltransferases Form Metabolite-Dependent, Covalent Adducts with Previously Methylated RNA. Biochemical 2014, 53, 7132–7144. [Google Scholar] [CrossRef]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-Mediated Cytosine-5 Methylation of Vault Noncoding RNA Determines Its Processing into Regulatory Small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef]
- Stojković, V.; Chu, T.; Therizols, G.; Weinberg, D.E.; Fujimori, D.G. miCLIP-MaPseq, a Substrate Identification Approach for Radical SAM RNA Methylating Enzymes. J. Am. Chem. Soc. 2018, 140, 7135–7143. [Google Scholar] [CrossRef]
- Hussain, S. Catalytic Crosslinking-Based Methods for Enzyme-Specified Profiling of RNA Ribonucleotide Modifications. Methods 2019, 156, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Khoddami, V.; Cairns, B.R. Identification of Direct Targets and Modified Bases of RNA Cytosine Methyltransferases. Nat. Biotechnol. 2013, 31, 458–464. [Google Scholar] [CrossRef]
- Khoddami, V.; Cairns, B.R. Transcriptome-Wide Target Profiling of RNA Cytosine Methyltransferases Using the Mechanism-Based Enrichment Procedure Aza-IP. Nat. Protoc. 2014, 9, 337–361. [Google Scholar] [CrossRef] [PubMed]
- Khoddami, V.; Yerra, A.; Cairns, B.R. Experimental Approaches for Target Profiling of RNA Cytosine Methyltransferases. Methods Enzymol. 2015, 560, 273–296. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Santi, D.V. Covalent Adducts between tRNA (m5U54)-Methyltransferase and RNA Substrates. Biochemical 1992, 31, 10295–10302. [Google Scholar] [CrossRef]
- Kealey, J.T.; Santi, D.V. Stereochemistry of tRNA(m5U54)-Methyltransferase Catalysis: 19F NMR Spectroscopy of an Enzyme-FUraRNA Covalent Complex. Biochemical 1995, 34, 2441–2446. [Google Scholar] [CrossRef]
- Carter, J.-M.; Emmett, W.; Mozos, I.R.; Kotter, A.; Helm, M.; Ule, J.; Hussain, S. FICC-Seq: A Method for Enzyme-Specified Profiling of Methyl-5-Uridine in Cellular RNA. Nucleic Acids Res. 2019, 47, e113. [Google Scholar] [CrossRef]
- Meyer, K.D. DART-seq: An Antibody-Free Method for Global m6a Detection. Nat. Methods 2019, 16, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Grozhik, A.V.; Jaffrey, S.R. Distinguishing RNA Modifications from Noise in Epitranscriptome Maps. Nat. Chem. Biol. 2018, 14, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Grozhik, A.V.; Jaffrey, S.R. Shrinking Maps of RNA Modifications. Nat. Cell Biol. 2017, 551, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Sas-Chen, A.; Schwartz, S. Misincorporation Signatures for Detecting Modifications in mRNA: Not as Simple as It Sounds. Methods 2019, 156, 53–59. [Google Scholar] [CrossRef]
- Schwartz, S. m1A Within Cytoplasmic mRNAs at Single Nucleotide Resolution: A Reconciled Transcriptome-Wide Map. RNA 2018, 24, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Li, X.; Wang, K.; Yi, C. Perspectives on Topology of the Human m1A Methylome at Single Nucleotide Resolution. RNA 2018, 24, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Diroma, M.A.; Ciaccia, L.; Pesole, G.; Picardi, E. Elucidating the Editome: Bioinformatics Approaches for RNA Editing Detection. Briefings Bioinform. 2019, 20, 436–447. [Google Scholar] [CrossRef]
- Parker, B.J. Statistical Methods for Transcriptome-Wide Analysis of RNA Methylation by Bisulfite Sequencing. In Advanced Structural Safety Studies; Springer International Publishing: Berlin, Germany, 2017; Volume 1562, pp. 155–167. [Google Scholar]
- Hauenschild, R.; Werner, S.; Tserovski, L.; Hildebrandt, A.; Motorin, Y.; Helm, M. CoverageAnalyzer (CAn): A Tool for Inspection of Modification Signatures in RNA Sequencing Profiles. Biomolecules 2016, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Piechotta, M.; Wyler, E.; Ohler, U.; Landthaler, M.; Dieterich, C. JACUSA: Site-Specific Identification of RNA Editing Events from Replicate Sequencing Data. BMC Bioinform. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Busan, S.; Weeks, K.M. Accurate Detection of Chemical Modifications in RNA by Mutational Profiling (MaP) with ShapeMapper 2. RNA 2018, 24, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Bormann, F.; Tuorto, F.; Cirzi, C.; Lyko, F.; Legrand, C. BisAMP: A Web-Based Pipeline for Targeted RNA Cytosine-5 Methylation Analysis. Methods 2019, 156, 121–127. [Google Scholar] [CrossRef]
- Xu, L.; Seki, M. Recent Advances in the Detection of Base Modifications Using the Nanopore Sequencer. J. Hum. Genet. 2020, 65, 25–33. [Google Scholar] [CrossRef]
- Vilfan, I.D.; Tsai, Y.-C.; A Clark, T.; Wegener, J.; Dai, Q.; Yi, C.; Pan, T.; Turner, S.W.; Korlach, J. Analysis of RNA Base Modification and Structural Rearrangement by Single-Molecule Real-Time Detection of Reverse Transcription. J. Nanobiotechnol. 2013, 11, 8. [Google Scholar] [CrossRef]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate Detection of m6A RNA Modifications in Native RNA Sequences. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading Canonical and Modified Nucleobases in 16S Ribosomal RNA Using Nanopore Native RNA Sequencing. PLoS ONE 2019, 14, e0216709. [Google Scholar] [CrossRef]
- Lorenz, D.A.; Sathe, S.; Einstein, J.M.; Yeo, G.W. Direct RNA Sequencing Enables m6A Detection in Endogenous Transcript Isoforms at Base-Specific Resolution. RNA 2020, 26, 19–28. [Google Scholar] [CrossRef]
- Jenjaroenpun, P.; Wongsurawat, T.; Wadley, T.D.; Wassenaar, T.M.; Liu, J.; Dai, Q.; Wanchai, V.; Akel, N.S.; Jamshidi-Parsian, A.; Franco, A.T.; et al. Decoding the Epitranscriptional Landscape from Native RNA Sequences. Nucleic Acids Res. 2021, 49, e7. [Google Scholar] [CrossRef]
- Ding, H.; Bailey, A.D.; Jain, M.; Olsen, H.; Paten, B. Gaussian Mixture Model-Based Unsupervised Nucleotide Modification Number Detection Using Nanopore-Sequencing Readouts. Bioinformatics 2020, 36, 4928–4934. [Google Scholar] [CrossRef]
- Cozzuto, L.; Liu, H.; Pryszcz, L.P.; Pulido, T.H.; Delgado-Tejedor, A.; Ponomarenko, J.; Novoa, E.M. MasterOfPores: A Workflow for the Analysis of Oxford Nanopore Direct RNA Sequencing Datasets. Front. Genet. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA Nanopore Sequencing of Full-Length Coronavirus Genomes Provides Novel Insights into Structural Variants and Enables Modification Analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motorin, Y.; Marchand, V. Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update. Genes 2021, 12, 278. https://doi.org/10.3390/genes12020278
Motorin Y, Marchand V. Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update. Genes. 2021; 12(2):278. https://doi.org/10.3390/genes12020278
Chicago/Turabian StyleMotorin, Yuri, and Virginie Marchand. 2021. "Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update" Genes 12, no. 2: 278. https://doi.org/10.3390/genes12020278
APA StyleMotorin, Y., & Marchand, V. (2021). Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update. Genes, 12(2), 278. https://doi.org/10.3390/genes12020278