
BARD1 Pathogenic Variants Are Associated with Triple-Negative Breast Cancer in a Spanish Hereditary Breast and Ovarian Cancer Cohort

 , , , , ,
, , , , ,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Controls
2.2. NGS Panel Testing
2.3. Variant Nomenclature
2.4. Co-Segregation Analysis and Loss of Heterozygosity (LOH)
2.5. gnomAD Analysis
2.6. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Brakeleer, S.; De Grève, J.; Loris, R.; Janin, N.; Lissens, W.; Sermijn, E.; Teugels, E. Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Hum. Mutat. 2010, 31, e1175–e1185. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Pharoah, P.D.P.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.W.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.; Le Trong, I.; Rajagopal, P.; Brzovic, P.S.; Stenkamp, R.E.; Klevit, R.E. Crystal structure of the BARD1 ankyrin repeat domain and its functional consequences. J. Biol. Chem. 2008, 283, 21179–21186. [Google Scholar] [CrossRef]
- Birrane, G.; Varma, A.K.; Soni, A.; Ladias, J.A.A. Crystal structure of the BARD1 BRCT domains. Biochemistry 2007, 46, 7706–7712. [Google Scholar] [CrossRef]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasint, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Feki, A.; Jefford, C.E.; Berardi, P.; Wu, J.Y.; Cartier, L.; Krause, K.H.; Irminger-Finger, I. BARD1 induces apoptosis by catalysing phosphorylation of p53 by DNA-damage response kinase. Oncogene 2005, 24, 3726–3736. [Google Scholar] [CrossRef]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef]
- Slavin, T.P.; Maxwell, K.N.; Lilyquist, J.; Vijai, J.; Neuhausen, S.L.; Hart, S.N.; Ravichandran, V.; Thomas, T.; Maria, A.; Villano, D.; et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. npj Breast Cancer 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.; Borde, J.; Weber-Lassalle, K.; Horváth, J.; Niederacher, D.; Arnold, N.; Kaulfuß, S.; Ernst, C.; Paul, V.G.; Honisch, E.; et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res. 2019, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- De Brakeleer, S.; De Grève, J.; Desmedt, C.; Joris, S.; Sotiriou, C.; Piccart, M.; Pauwels, I.; Teugels, E. Frequent incidence of BARD1-truncating mutations in germline DNA from triple-negative breast cancer patients. Clin. Genet. 2016, 89, 336–340. [Google Scholar] [CrossRef] [PubMed]
- González-Rivera, M.; Lobo, M.; López-Tarruella, S.; Jerez, Y.; del Monte-Millán, M.; Massarrah, T.; Ramos-Medina, R.; Ocaña, I.; Picornell, A.; Garzón, S.S.; et al. Frequency of germline DNA genetic findings in an unselected prospective cohort of triple-negative breast cancer patients participating in a platinum-based neoadjuvant chemotherapy trial. Breast Cancer Res. Treat. 2016, 156, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J. Natl. Cancer Inst. 2018, 110, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Castéra, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018, 20, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers with Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2019, 5, 51–57. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef]
- Obón-Santacana, M.; Vilardell, M.; Carreras, A.; Duran, X.; Velasco, J.; Galván-Femenía, I.; Alonso, T.; Puig, L.; Sumoy, L.; Duell, E.J.; et al. GCAT|Genomes for life: A prospective cohort study of the genomes of Catalonia. BMJ Open 2018, 8, 18324. [Google Scholar] [CrossRef]
- Castellanos, E.; Gel, B.; Rosas, I.; Tornero, E.; Santín, S.; Pluvinet, R.; Velasco, J.; Sumoy, L.; Del Valle, J.; Perucho, M.; et al. A comprehensive custom panel design for routine hereditary cancer testing: Preserving control, improving diagnostics and revealing a complex variation landscape. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cabrera, J.M.; del Valle, J.; Castellanos, E.; Feliubadaló, L.; Pineda, M.; Brunet, J.; Serra, E.; Capellà, G.; Lázaro, C.; Gel, B. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur. J. Hum. Genet. 2020, 28. [Google Scholar] [CrossRef]
- Feliubadaló, L.; López-Fernández, A.; Pineda, M.; Díez, O.; del Valle, J.; Gutiérrez-Enríquez, S.; Teulé, A.; González, S.; Stjepanovic, N.; Salinas, M.; et al. Opportunistic testing of BRCA1, BRCA2 and mismatch repair genes improves the yield of phenotype driven hereditary cancer gene panels. Int. J. Cancer 2019, 145, 2682–2691. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; Mcgowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581. [Google Scholar] [CrossRef] [PubMed]
- Bonache, S.; Esteban, I.; Moles-Fernández, A.; Tenés, A.; Duran-Lozano, L.; Montalban, G.; Bach, V.; Carrasco, E.; Gadea, N.; López-Fernández, A.; et al. Multigene panel testing beyond BRCA1/2 in breast/ovarian cancer Spanish families and clinical actionability of findings. J. Cancer Res. Clin. Oncol. 2018, 144, 2495–2513. [Google Scholar] [CrossRef]
- Feliubadaló, L.; Tonda, R.; Gausachs, M.; Trotta, J.R.; Castellanos, E.; López-Doriga, A.; Teulé, À.; Tornero, E.; Del Valle, J.; Gel, B.; et al. Benchmarking of whole exome sequencing and Ad Hoc designed panels for genetic testing of hereditary cancer. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Suszynska, M.; Kozlowski, P. Summary of bard1 mutations and precise estimation of breast and ovarian cancer risks associated with the mutations. Genes (Basel) 2020, 11, 798. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Adedokun, B.; Zheng, Y.; Ndom, P.; Gakwaya, A.; Makumbi, T.; Zhou, A.Y.; Yoshimatsu, T.F.; Rodriguez, A.; Madduri, R.K.; Foster, I.T.; et al. Prevalence of inherited mutations in breast cancer predisposition genes among women in Uganda and Cameroon. Cancer Epidemiol. Biomarkers Prev. 2020, 29, 359–367. [Google Scholar] [CrossRef]
- Zeng, C.; Guo, X.; Wen, W.; Shi, J.; Long, J.; Cai, Q.; Shu, X.O.; Xiang, Y.; Zheng, W. Evaluation of pathogenetic mutations in breast cancer predisposition genes in population-based studies conducted among Chinese women. Breast Cancer Res. Treat. 2020, 181, 465–473. [Google Scholar] [CrossRef]
- Kaneyasu, T.; Mori, S.; Yamauchi, H.; Ohsumi, S.; Ohno, S.; Aoki, D.; Baba, S.; Kawano, J.; Miki, Y.; Matsumoto, N.; et al. Prevalence of disease-causing genes in Japanese patients with BRCA1/2-wildtype hereditary breast and ovarian cancer syndrome. npj Breast Cancer 2020, 6. [Google Scholar] [CrossRef]
- Carter, N.J.; Marshall, M.L.; Susswein, L.R.; Zorn, K.K.; Hiraki, S.; Arvai, K.J.; Torene, R.I.; McGill, A.K.; Yackowski, L.; Murphy, P.D.; et al. Germline pathogenic variants identified in women with ovarian tumors. Gynecol. Oncol. 2018, 151, 481–488. [Google Scholar] [CrossRef]
- Kwong, A.; Shin, V.Y.; Chen, J.; Cheuk, I.W.Y.; Ho, C.Y.S.; Au, C.H.; Chan, K.K.L.; Ngan, H.Y.S.; Chan, T.L.; Ford, J.M.; et al. Germline mutation in 1338 BRCA-negative Chinese hereditary breast and/or ovarian cancer patients: Clinical testing with a multigene test panel. J. Mol. Diagn. 2020, 22, 544–554. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Fierheller, C.T.; Recio, N.; Tonin, P.N. Literature review of BARD1 as a cancer predisposing gene with a focus on breast and ovarian cancers. Genes (Basel) 2020, 11, 856. [Google Scholar] [CrossRef]


{kind=link}
| Clinical Indication | Number of Patients (%) | Genes Tested by Phenotype | Number of PVs (%) | BARD1 (%) | BARD1 Excluding Patients with Other PVs (%) |
|---|---|---|---|---|---|
| Only Hereditary Breast Cancer, HBC | 2622 (65.31%) | ATM, BRCA1, BRCA2, CHEK2, MLH1, MSH2, MSH6, PALB2, TP53 | 270 PVs (10.30%): ATM (34), BRCA1 (71), BRCA2 (90), CHEK2 (27), MLH1 (3), MSH2 (1), MSH6 (2), PALB2 (37), TP53 (5) | 13 (0.50%) OR = 4.18 (2.10–7.70) **p = 5.45 × 10−5 | 13 (0.50%) OR = 4.18 (2.10–7.70) **p = 5.45 × 10−5 |
| Only Hereditary Ovarian Cancer, HOC | 715 (17.81%) | BRCA1, BRCA2, BRIP1, MLH1, MSH2, MSH6, RAD51C, RAD51D | 93 PVs (13.01%): BRCA1 (39), BRCA2 (35), BRIP1 (6), MLH1 (1), MSH6 (4), RAD51C (4), RAD51D (4) | 3 (0.42%) OR = 3.53 (0.71–10.86) p = 0.06 | 3 (0.42%) OR = 3.53 (0.71–10.86) p = 0.06 |
| Hereditary Breast and Ovarian Cancer, HBOC | 608 (15.14%) | ATM, BRCA1, BRCA2, BRIP1, CHEK2, MLH1, MSH2, MSH6, PALB2, RAD51C, RAD51D, TP53 | 104 PVs (17.11%): ATM (7), BRCA1 (45), BRCA2 (32), BRIP1 (7), CHEK2 (6), MSH2 (1), PALB2 (3), RAD51C (1), RAD51D (1), TP53 (1) | 3 (0.49%) OR = 4.16 (0.83–12.79) * p = 0.04 | 2 (0.33%) OR = 2.77 (0.33–10.47) p = 0.17 |
| HBC/HOC/HBOC + Other clinical indications | 70 (1.74%) | Details in Ref: [24] | 9 PVs (12.86%): ATM (2), BRCA1 (2), BRCA2 (1), MSH6 (2), PTEN (1), RAD51C (1) | 0 (0%) | 0 (0%) |
| Total | 4015 | 476 (11.86%) | 19 (0.47%) OR = 3.99 (2.25–6.77) ** p = 3.48 × 10−6 | 18 (0.45%) OR = 3.78 (2.10–6.48) ** p = 1.16 × 10−5 | |
| Controls studied | |||||
| Spanish population cohort (n = 194) | 0 (0%) | ||||
| gnomAD non-Finnish European, non-cancer cohort (n = 51,202) | 61 (0.12%) | ||||
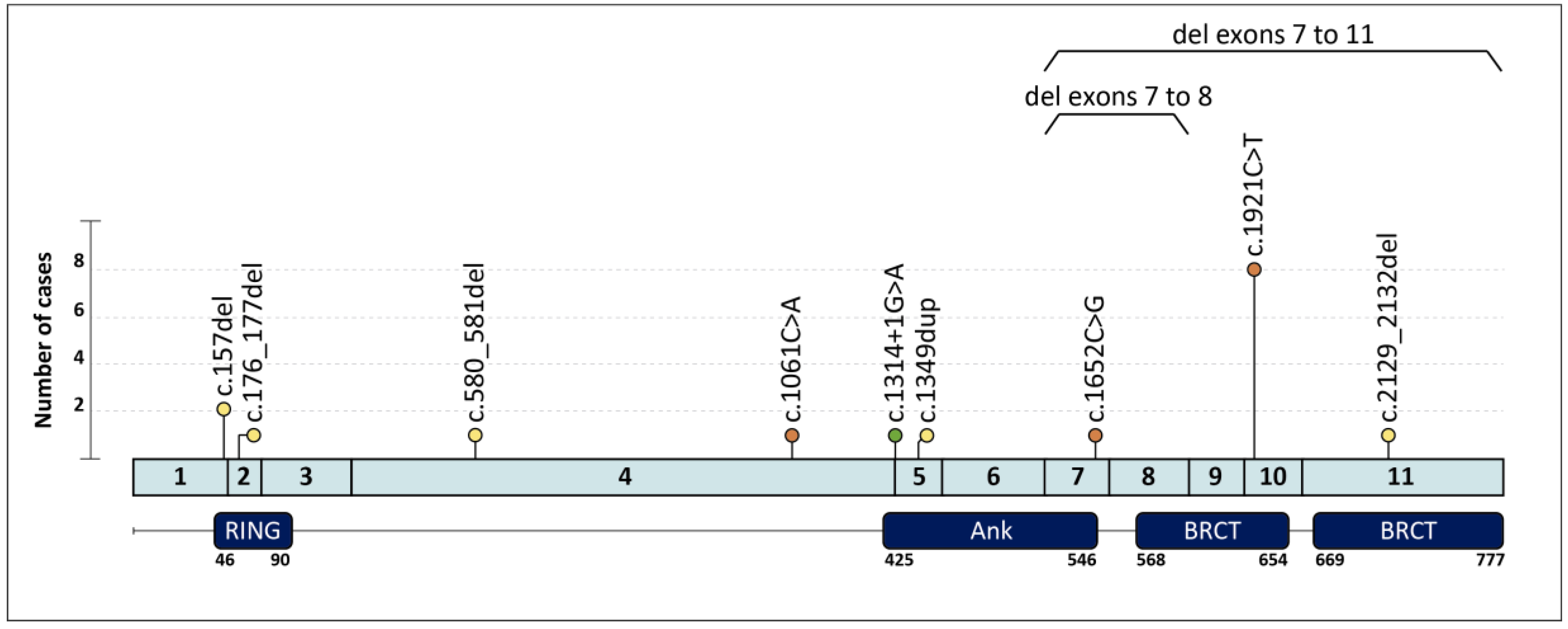
| Family | Clinical Indication | Cancer Type (Age at dx) | Tumor Phenotype | Family History (Age at dx) | BARD1 PV (c.) | BARD1 PV (p.) | Additional PVs |
|---|---|---|---|---|---|---|---|
| 1 | HBC | Breast (40,58) | ILC ER+ Her2-; TNBC | Cousin: PC (73) | c.157del | p.(Cys53Valfs*5) | |
| 2 | HBOC | Breast (30) | IDC ER+ Her2- | ||||
| 3 | HOC | Ovary (59) | HGOSC | c.176_177del | p.(Glu59Alafs*8) | ||
| 4 † | HBC | Breast (27,42) | ER+ BC; TNBC | Mother: Breast (44,44) | c.580_581del | p.(Arg194Glyfs*2) | |
| 5 | HBC | Breast (38) | IDC ER+ Her2- | Aunt: Breast (37) ‡, Aunt: Breast (36) | c.1061C > A | p.(Ser354*) | |
| 6 | HOC | Ovary (62) | EC | c.1314+1G > A | p.? | ||
| 7 | HBC | Breast (49) | TNBC | c.1349dup | p.(Asn450Lysfs*4) | ||
| 8 | HBC | Breast (31) | TNBC | Aunt: Breast (64) ‡, Aunt: Breast (64) ‡ | c.1652C > G | p.(Ser551*) | |
| 9 | HBC | Breast (56) | TNBC | c.1921C > T | p.(Arg641*) | ||
| 10 | HBOC | Breast (54) | IDC ER+ Her2- | Mother: Ovary (63) | BRCA2 c.3264dupT; p.(Gln1089Serfs*10) | ||
| 11 | HBC | Breast (63) | TNBC | Aunt: Breast (60) ‡, Cousin: Breast (54) ‡ | |||
| 12 | HBC | Breast (40) | IDC ER+ Her2+ | Mother: EC (62), Breast (64) | |||
| 13 | HBC | Breast (49) | TNBC | ||||
| 14 | HBC | Breast (30) | TNBC | ||||
| 15 | HBC | Breast (46,56,56) | IDBC; bilateral IDBC | Mother: Breast (78) | |||
| 16 ^ | HBC | Breast (40,47) | TNBC; TNBC | Sister: Breast (46); Sister: Breast (48); Mother: Breast (48); Cousin: Breast (46) | |||
| 17 | HBOC | Breast (42) | IDC ER+ Her2- | Uncle: Breast (71); Aunt: Ovary (62) | c.2129_2132del | p.(Asp710Valfs*3) | |
| 18 | HOC | Ovary (62) | HGOSC | c.(1568+1_1569-1)_(1810+1_1811-1)del Exons 7–8 deletion | |||
| 19 | HBC | Breast (44) | TNBC | Mother: Breast (69); Aunt: Breast (60) | g.(?_215617227)_(215593730_?) Exons 7–11 deletion |
| Group | Number of Patients | BARD1-Mutated |
|---|---|---|
| TNBC patients | 680 | 10 (0.88%) OR = 5.40 (1.77–18.15) p = 0.001 ** |
| Non-TNBC patients | 2179 | 6 (0.28%) |
| Total | 2859 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rofes, P.; Del Valle, J.; Torres-Esquius, S.; Feliubadaló, L.; Stradella, A.; Moreno-Cabrera, J.M.; López-Doriga, A.; Munté, E.; De Cid, R.; Campos, O.; et al. BARD1 Pathogenic Variants Are Associated with Triple-Negative Breast Cancer in a Spanish Hereditary Breast and Ovarian Cancer Cohort. Genes 2021, 12, 150. https://doi.org/10.3390/genes12020150
Rofes P, Del Valle J, Torres-Esquius S, Feliubadaló L, Stradella A, Moreno-Cabrera JM, López-Doriga A, Munté E, De Cid R, Campos O, et al. BARD1 Pathogenic Variants Are Associated with Triple-Negative Breast Cancer in a Spanish Hereditary Breast and Ovarian Cancer Cohort. Genes. 2021; 12(2):150. https://doi.org/10.3390/genes12020150
Chicago/Turabian StyleRofes, Paula, Jesús Del Valle, Sara Torres-Esquius, Lídia Feliubadaló, Agostina Stradella, José Marcos Moreno-Cabrera, Adriana López-Doriga, Elisabet Munté, Rafael De Cid, Olga Campos, and et al. 2021. "BARD1 Pathogenic Variants Are Associated with Triple-Negative Breast Cancer in a Spanish Hereditary Breast and Ovarian Cancer Cohort" Genes 12, no. 2: 150. https://doi.org/10.3390/genes12020150
APA StyleRofes, P., Del Valle, J., Torres-Esquius, S., Feliubadaló, L., Stradella, A., Moreno-Cabrera, J. M., López-Doriga, A., Munté, E., De Cid, R., Campos, O., Cuesta, R., Teulé, Á., Grau, È., Sanz, J., Capellá, G., Díez, O., Brunet, J., Balmaña, J., & Lázaro, C. (2021). BARD1 Pathogenic Variants Are Associated with Triple-Negative Breast Cancer in a Spanish Hereditary Breast and Ovarian Cancer Cohort. Genes, 12(2), 150. https://doi.org/10.3390/genes12020150