The Novel Halovirus Hardycor1, and the Presence of Active (Induced) Proviruses in Four Haloarchaea



Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Isolation
2.2. DNA Isolation, Sequencing and Assembly
2.3. Bioinformatics Analyses
3. Results
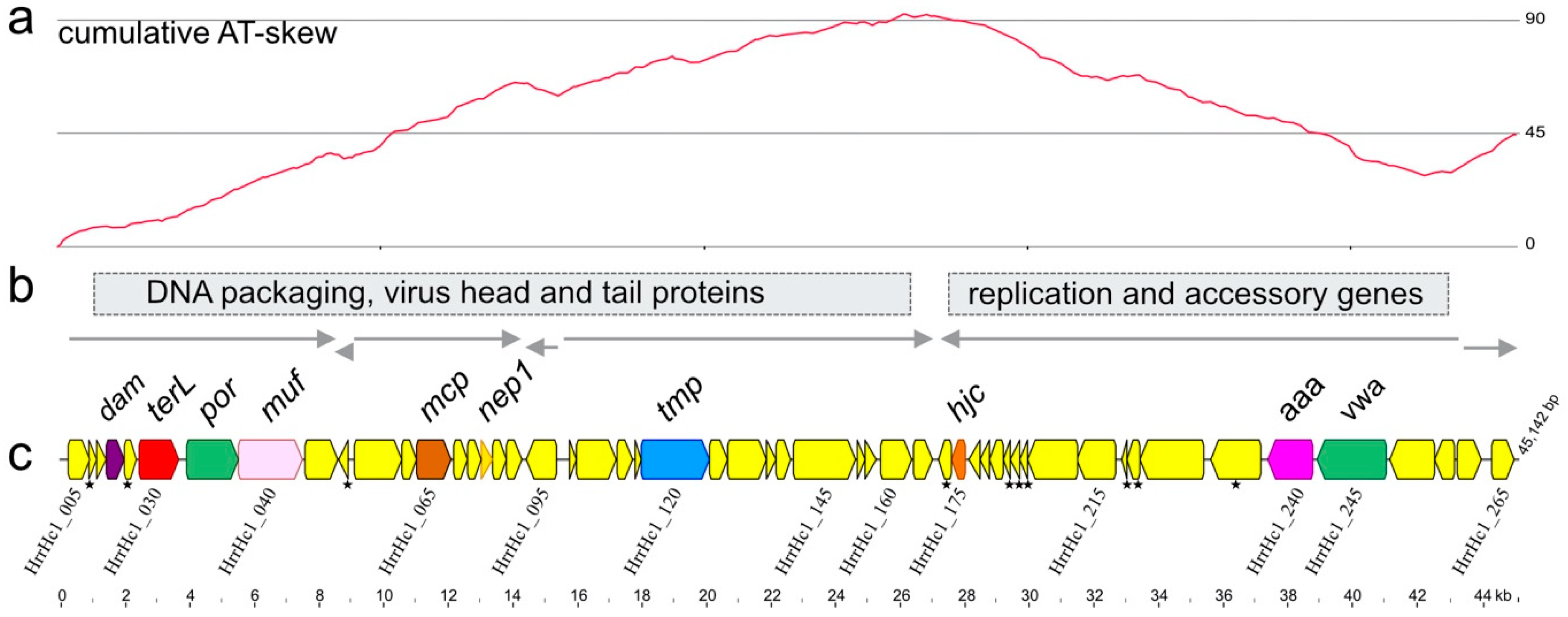
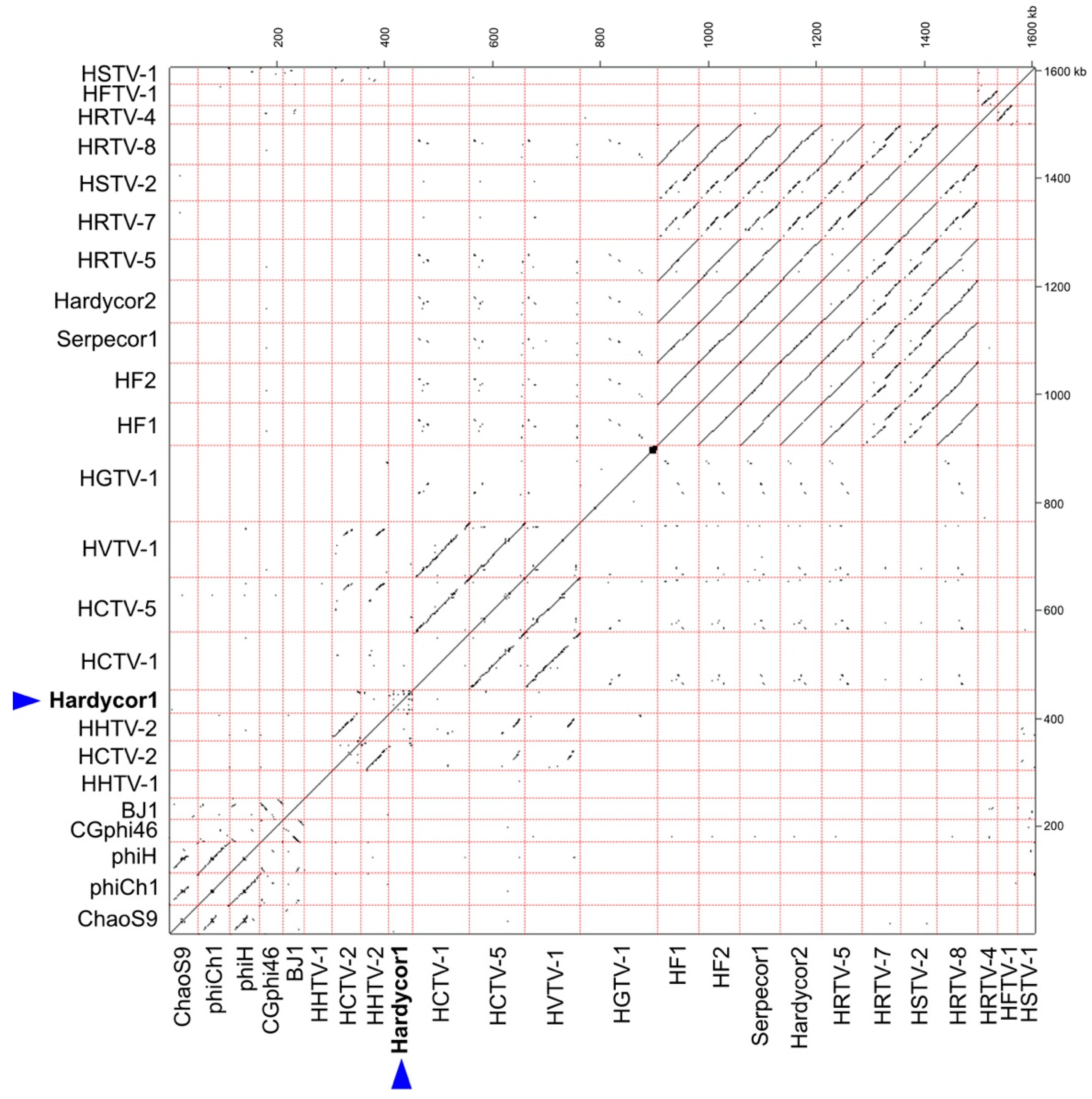
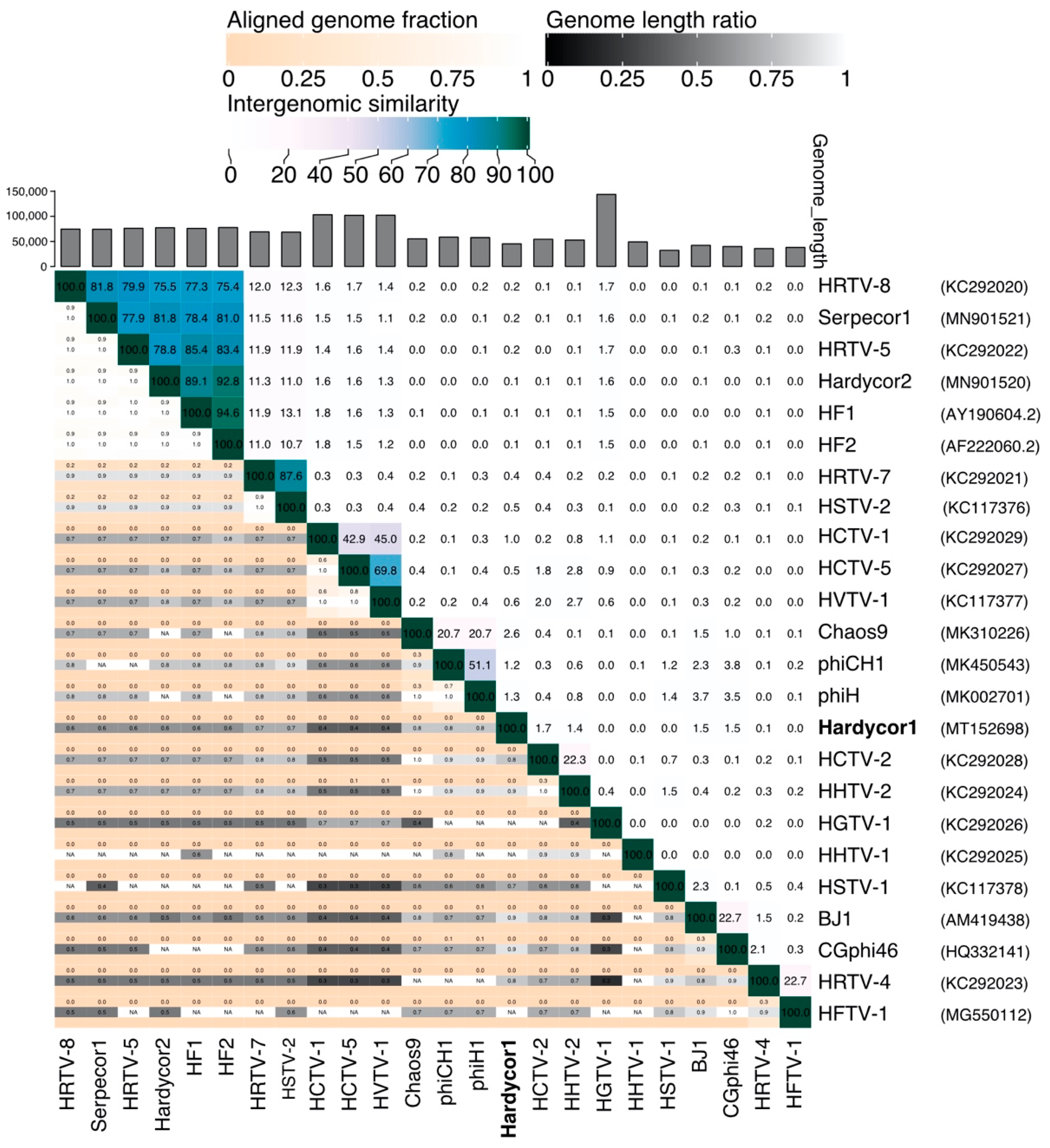
3.1. Isolation and Sequence
3.2. Annotation and Predicted Proteins
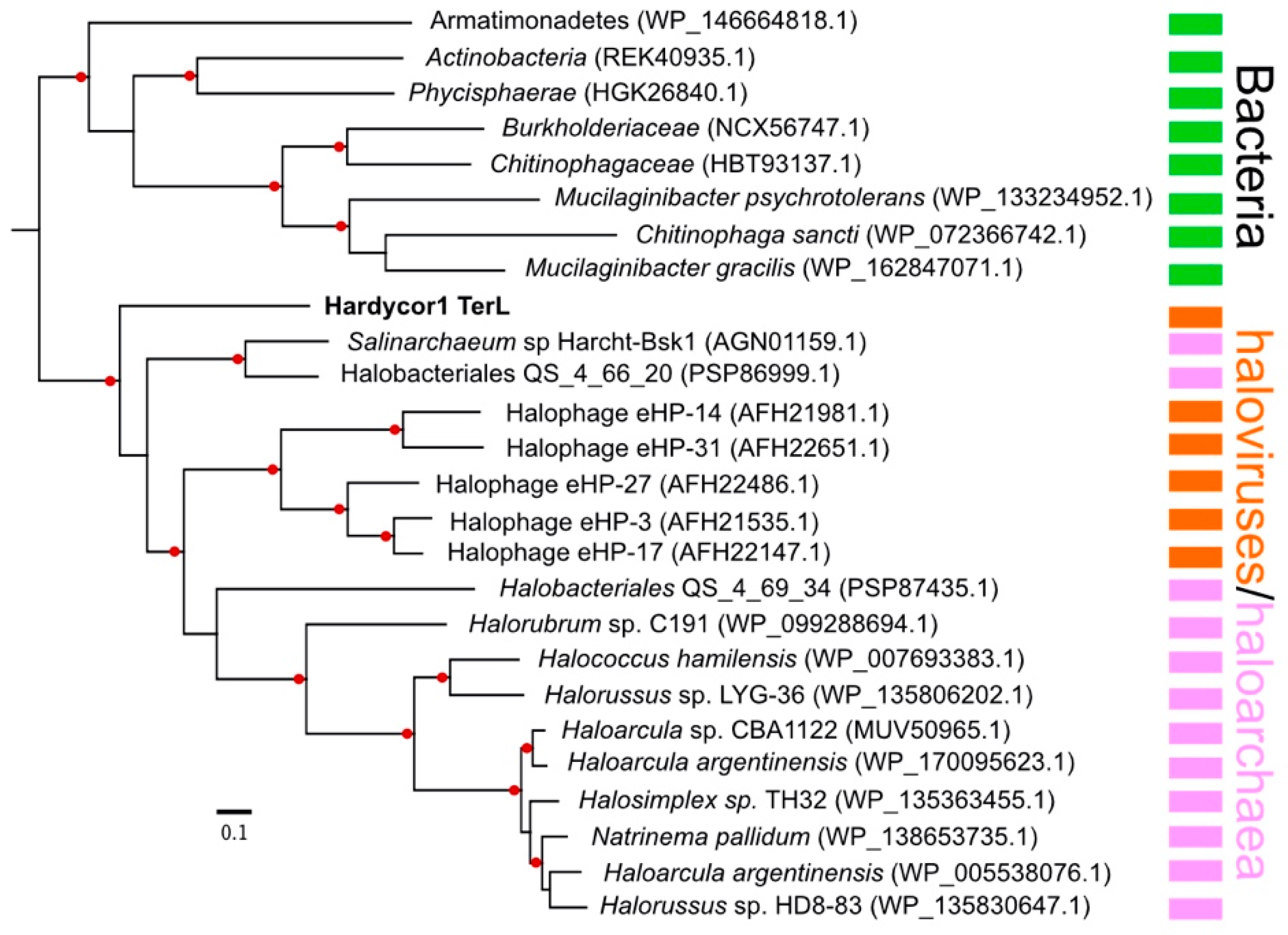
3.3. Protein-Based Phylogenetic Analyses
3.4. Match to CRISPR Spacer
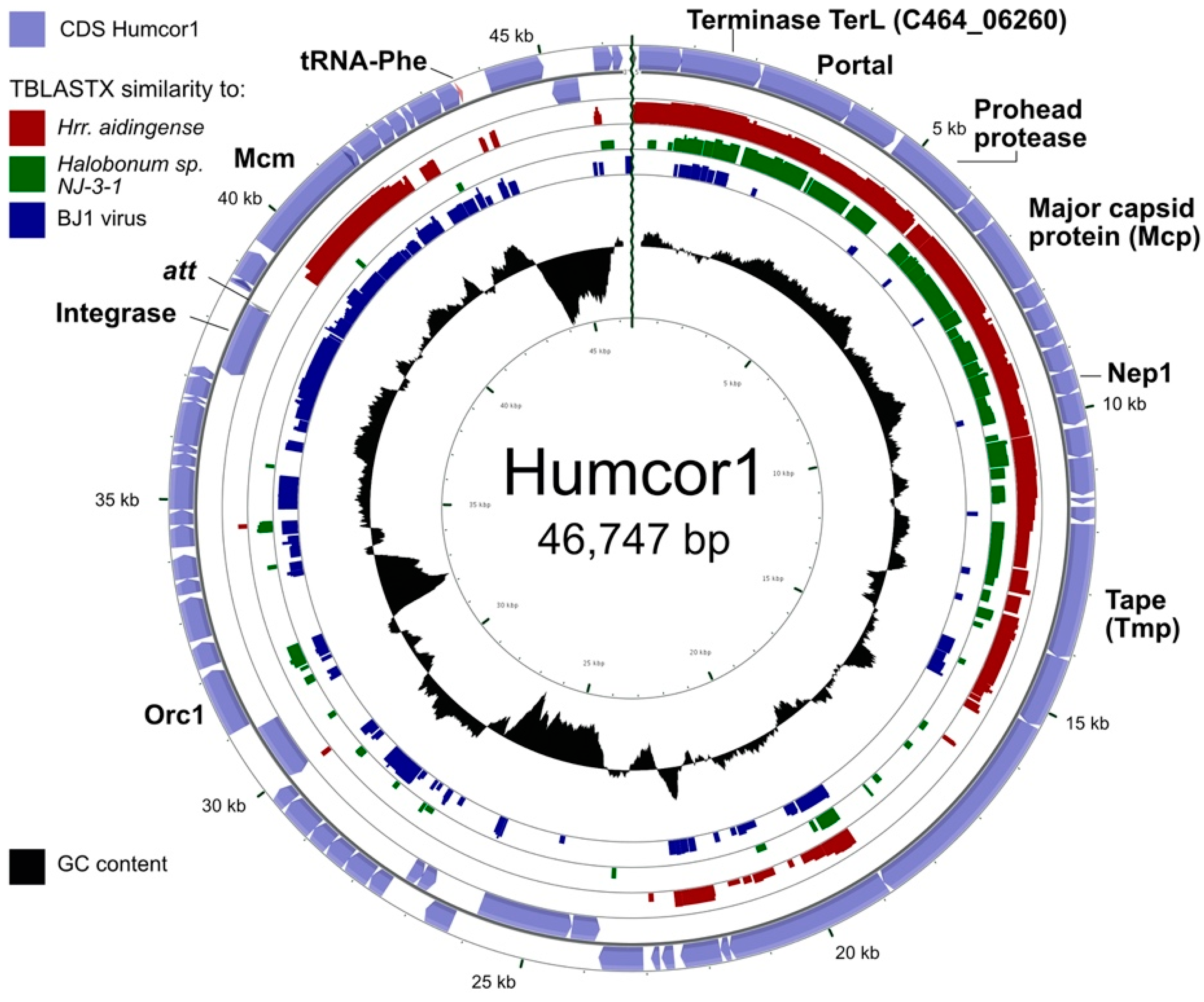
3.5. Active Proviruses of Hrr. coriense
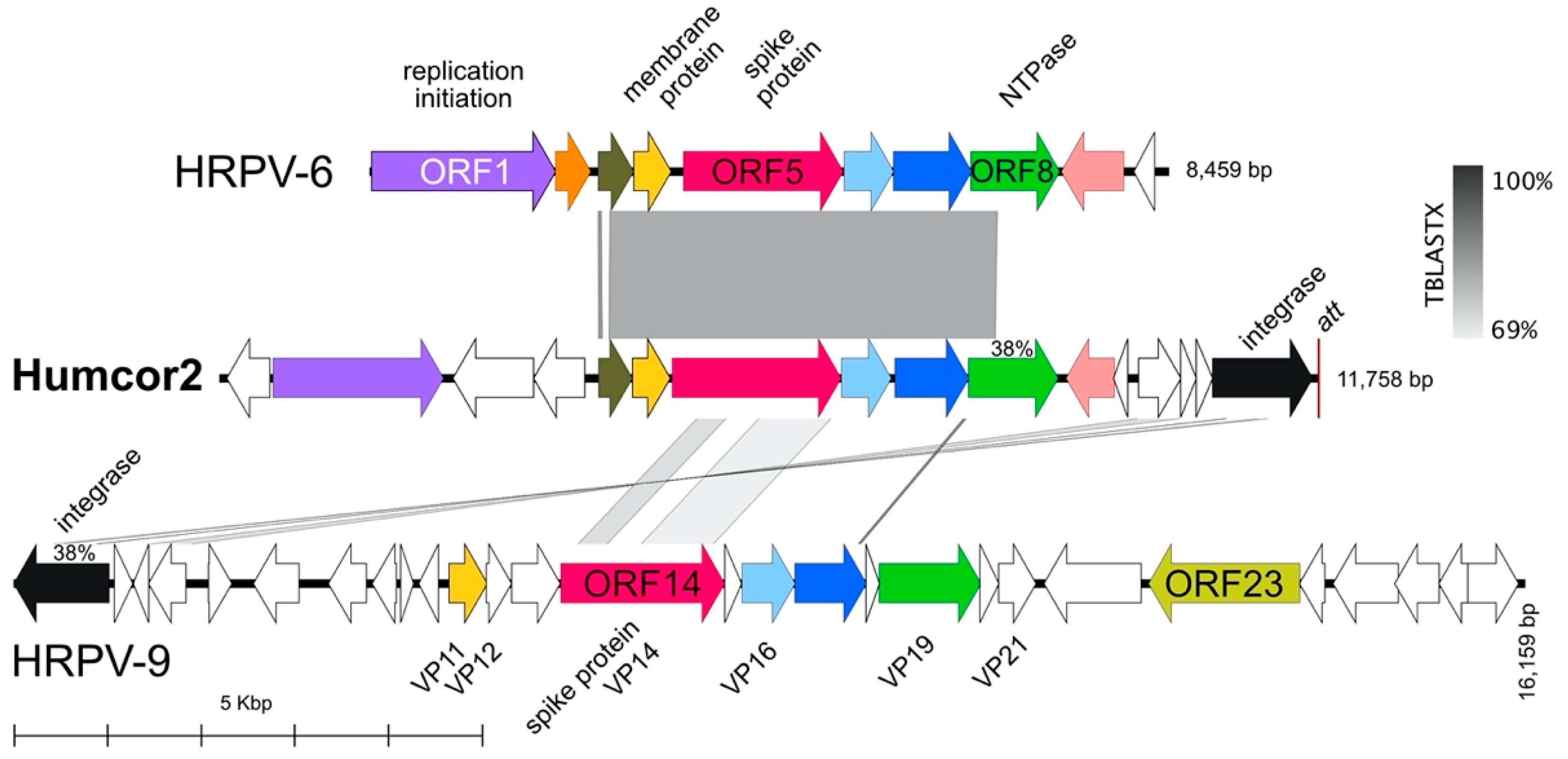
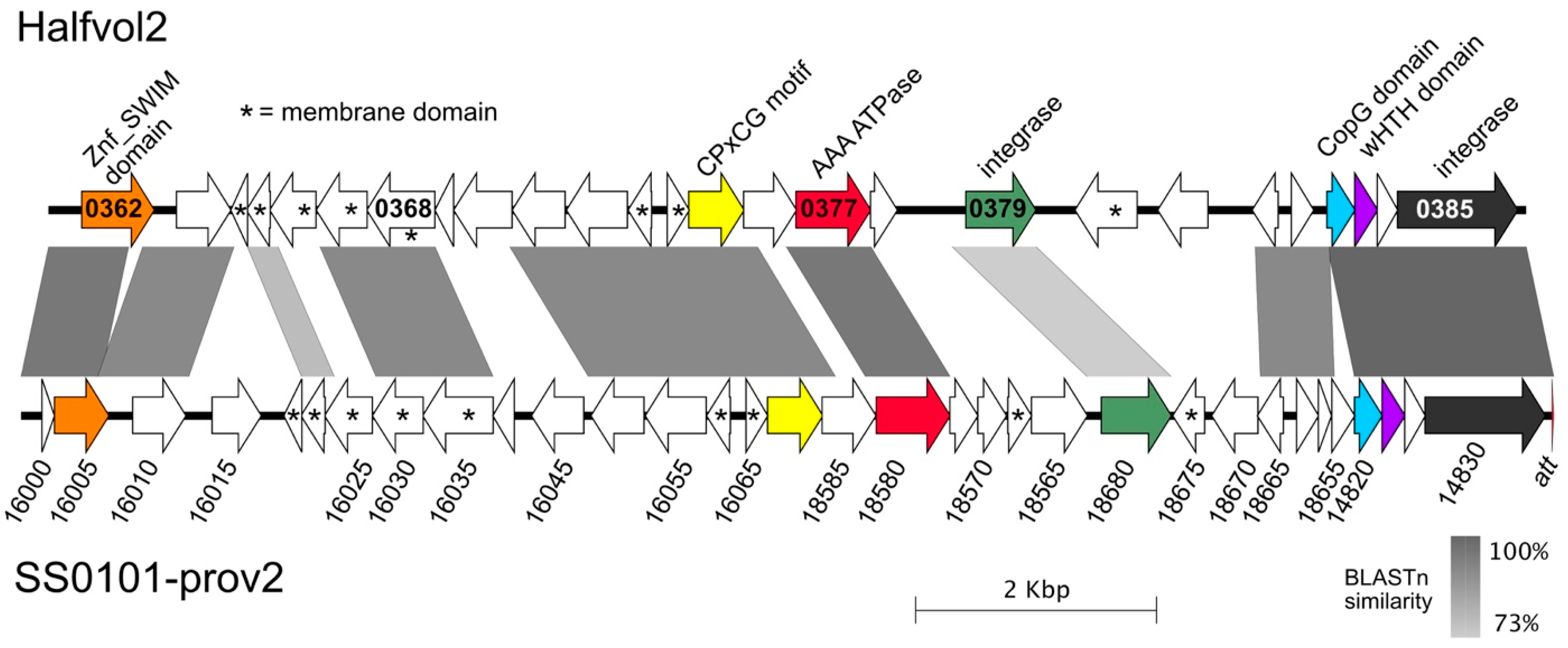
3.6. Proviruses Present in Virus Stocks from Other Haloarchaeal Hosts
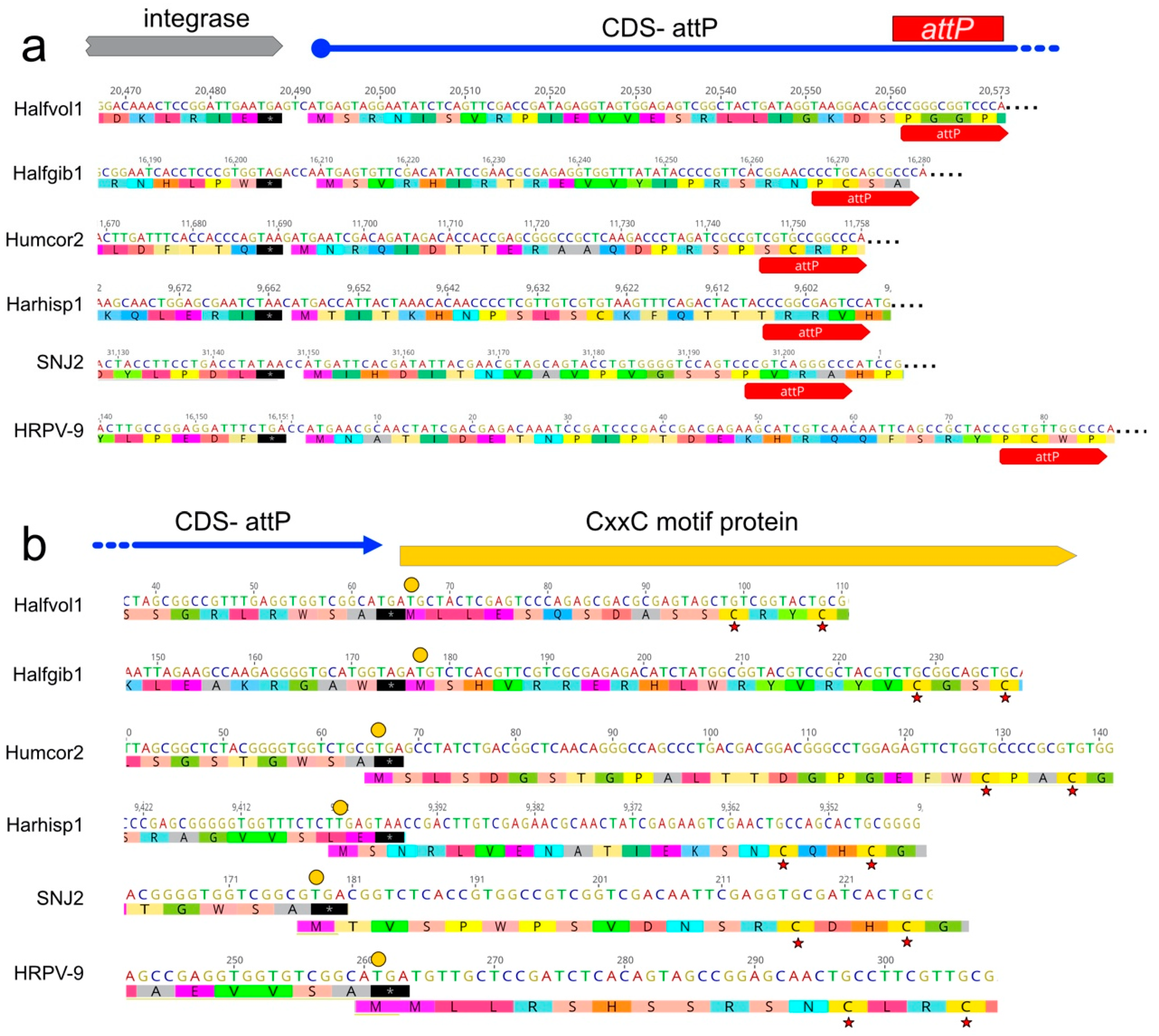
3.7. A CDS Frequently Encompasses the attP Sequence of Pleolipovirus-Like Proviruses
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bergh, O.; Borsheim, K.Y.; Bratbak, G.; Heldal, M. High abundance of viruses found in aquatic environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Hendrix, R. Bacteriophages: Evolution of the majority. Theor. Popul. Biol. 2002, 61, 471–480. [Google Scholar] [CrossRef]
- Coutinho, F.H.; Cabello-Yeves, P.J.; Gonzalez-Serrano, R.; Rosselli, R.; Lopez-Perez, M.; Zemskaya, T.I.; Zakharenko, A.S.; Ivanov, V.G.; Rodriguez-Valera, F. New viral biogeochemical roles revealed through metagenomic analysis of Lake Baikal. Microbiome 2020, 8, 163. [Google Scholar] [CrossRef]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015, 34, 169–183. [Google Scholar] [CrossRef]
- Isaev, A.; Drobiazko, A.; Sierro, N.; Gordeeva, J.; Yosef, I.; Qimron, U.; Ivanov, N.V.; Severinov, K. Phage T7 DNA mimic protein Ocr is a potent inhibitor of BREX defence. Nucleic Acids Res. 2020, 48, 7601–7602. [Google Scholar] [CrossRef]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis-lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef]
- Dyall-Smith, M.; Pfeifer, F.; Witte, A.; Oesterhelt, D.; Pfeiffer, F. Complete genome sequence of the model halovirus phih1 (φh1). Genes 2018, 9, 493. [Google Scholar] [CrossRef]
- Tang, S.L.; Nuttall, S.; Dyall-Smith, M. Haloviruses HF1 and HF2: Evidence for a recent and large recombination event. J. Bacteriol. 2004, 186, 2810–2817. [Google Scholar] [CrossRef]
- Krupovic, M.; Forterre, P.; Bamford, D.H. Comparative analysis of the mosaic genomes of tailed archaeal viruses and proviruses suggests common themes for virion architecture and assembly with tailed viruses of bacteria. J. Mol. Biol. 2010, 397, 144–160. [Google Scholar] [CrossRef]
- Pietila, M.K.; Laurinmaki, P.; Russell, D.A.; Ko, C.C.; Jacobs-Sera, D.; Butcher, S.J.; Bamford, D.H.; Hendrix, R.W. Insights into head-tailed viruses infecting extremely halophilic archaea. J. Virol. 2013, 87, 3248–3260. [Google Scholar] [CrossRef]
- Sencilo, A.; Jacobs-Sera, D.; Russell, D.A.; Ko, C.C.; Bowman, C.A.; Atanasova, N.S.; Osterlund, E.; Oksanen, H.M.; Bamford, D.H.; Hatfull, G.F.; et al. Snapshot of haloarchaeal tailed virus genomes. RNA Biol. 2013, 10, 803–816. [Google Scholar] [CrossRef]
- Sencilo, A.; Roine, E. A glimpse of the genomic diversity of haloarchaeal tailed viruses. Front. Microbiol. 2014, 5, 84. [Google Scholar]
- Krupovic, M.; Quemin, E.R.; Bamford, D.H.; Forterre, P.; Prangishvili, D. Unification of the globally-distributed spindle-shaped viruses of archaea. J. Virol. 2013, 2354–2358. [Google Scholar] [CrossRef]
- Pietila, M.K.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Modified coat protein forms the flexible spindle-shaped virion of haloarchaeal virus His1. Environ. Microbiol. 2013, 15, 1674–1686. [Google Scholar]
- Bath, C.; Dyall-Smith, M.L. His1, an archaeal virus of the Fuselloviridae family that infects Haloarcula hispanica. J. Virol. 1998, 72, 9392–9395. [Google Scholar] [CrossRef]
- Demina, T.A.; Oksanen, H.M. Pleomorphic archaeal viruses: The family Pleolipoviridae is expanding by seven new species. Arch. Virol. 2020, 165, 2723–2731. [Google Scholar] [CrossRef]
- Lee, S.T.M.; Ding, J.Y.; Chiang, P.W.; Dyall-Smith, M.; Tang, S.L. Insights into gene regulation of the halovirus His2 infecting Haloarcula hispanica. Microbiologyopen 2020, 9, e1016. [Google Scholar] [CrossRef]
- Porter, K.; Kukkaro, P.; Bamford, J.K.; Bath, C.; Kivela, H.M.; Dyall-Smith, M.L.; Bamford, D.H. SH1: A novel, spherical halovirus isolated from an australian hypersaline lake. Virology 2005, 335, 22–33. [Google Scholar] [CrossRef]
- Porter, K.; Russ, B.E.; Yang, J.; Dyall-Smith, M.L. The transcription programme of the protein-primed halovirus SH1. Microbiology 2008, 154, 3599–3608. [Google Scholar] [CrossRef]
- Nuttall, S.D.; Dyall-Smith, M.L. Halophage HF2: Genome organization and replication strategy. J. Virol. 1995, 69, 2322–2327. [Google Scholar] [CrossRef] [PubMed]
- Dyall-Smith, M.; Tang, S.L.; Russ, B.; Chiang, P.W.; Pfeiffer, F. Comparative genomics of two new HF1-like haloviruses. Genes 2020, 11, 405. [Google Scholar] [CrossRef] [PubMed]
- Dyall-Smith, M.L. The Halohandbook: Protocols for Halobacterial Genetics. Available online: http://www.haloarchaea.com/resources/halohandbook/ (accessed on 28 February 2020).
- Geneious. Available online: https://www.geneious.com/geneious/ (accessed on 1 December 2019).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lomsadze, A.; Gemayel, K.; Tang, S.; Borodovsky, M. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 2018, 28, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Noe, L.; Kucherov, G. Yass: Enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 2005, 33, W540–W543. [Google Scholar] [CrossRef] [PubMed]
- Yass Genomic Similarity Search Tool. Available online: http://bioinfo.lifl.fr/yass/index.php (accessed on 1 June 2020).
- Genewiz Browser. Available online: http://www.cbs.dtu.dk/services/gwBrowser/ (accessed on 1 June 2020).
- Img/vr Spacer Blast Tool. Available online: https://img.jgi.doe.gov/cgi-bin/vr), (accessed on 1 June 2020).
- CRISPRs Web Server. Available online: http://crispr.i2bc.paris-saclay.fr/ (accessed on 1 June 2020).
- VIRFAM, Remote Homology Detection of Viral Protein Families. Available online: http://biodev.cea.fr/virfam/ (accessed on 1 June 2020).
- Lopes, A.; Tavares, P.; Petit, M.A.; Guerois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genomics 2014, 15, 1027. [Google Scholar] [CrossRef]
- Phobius. A Combined Transmembrane Topology and Signal Peptide Predictor. Available online: https://phobius.sbc.su.se/ (accessed on 1 June 2020).
- Kall, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef]
- Becker, E.A.; Seitzer, P.M.; Tritt, A.; Larsen, D.; Krusor, M.; Yao, A.I.; Wu, D.; Madern, D.; Eisen, J.A.; Darling, A.E.; et al. Phylogenetically driven sequencing of extremely halophilic archaea reveals strategies for static and dynamic osmo-response. PLoS Genet. 2014, 10, e1004784. [Google Scholar] [CrossRef]
- Nuttall, S.D.; Dyall-Smith, M.L. Ch2, a novel halophilic archaeon from an australian solar saltern. Int. J. Syst. Bacteriol. 1993, 43, 729–734. [Google Scholar] [CrossRef][Green Version]
- REBASE. The Restriction Enzyme Database. Available online: http://rebase.neb.com/rebase/rebase.html (accessed on 1 June 2020).
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC-a novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar]
- Barylski, J.; Enault, F.; Dutilh, B.E.; Schuller, M.B.; Edwards, R.A.; Gillis, A.; Klumpp, J.; Knezevic, P.; Krupovic, M.; Kuhn, J.H.; et al. Analysis of spounaviruses as a case study for the overdue reclassification of tailed phages. Syst. Biol. 2020, 69, 110–123. [Google Scholar] [PubMed]
- Meier-Kolthoff, J.P.; Goker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [PubMed]
- Jamet, A.; Touchon, M.; Ribeiro-Goncalves, B.; Carrico, J.A.; Charbit, A.; Nassif, X.; Ramirez, M.; Rocha, E.P.C. A widespread family of polymorphic toxins encoded by temperate phages. BMC Biol. 2017, 15, 75. [Google Scholar]
- Xu, J.; Hendrix, R.W.; Duda, R.L. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol. Cell 2004, 16, 11–21. [Google Scholar]
- Xu, J.; Hendrix, R.W.; Duda, R.L. Chaperone-protein interactions that mediate assembly of the bacteriophage lambda tail to the correct length. J. Mol. Biol. 2014, 426, 1004–1018. [Google Scholar]
- Mahony, J.; Alqarni, M.; Stockdale, S.; Spinelli, S.; Feyereisen, M.; Cambillau, C.; Sinderen, D.V. Functional and structural dissection of the tape measure protein of lactococcal phage TP901-1. Sci. Rep. 2016, 6, 36667. [Google Scholar]
- Tebbe, A.; Klein, C.; Bisle, B.; Siedler, F.; Scheffer, B.; Garcia-Rizo, C.; Wolfertz, J.; Hickmann, V.; Pfeiffer, F.; Oesterhelt, D. Analysis of the cytosolic proteome of Halobacterium salinarum and its implication for genome annotation. Proteomics 2005, 5, 168–179. [Google Scholar] [CrossRef]
- Murphy, J.; Bottacini, F.; Mahony, J.; Kelleher, P.; Neve, H.; Zomer, A.; Nauta, A.; van Sinderen, D. Comparative genomics and functional analysis of the 936 group of lactococcal Siphoviridae phages. Sci. Rep. 2016, 6, 21345. [Google Scholar]
- Wyatt, H.D.; West, S.C. Holliday junction resolvases. Cold Spring Harb. Perspect. Biol. 2014, 6, a023192. [Google Scholar]
- Ennifar, E.; Basquin, J.; Birkenbihl, R.; Suck, D. Purification, crystallization and preliminary x-ray diffraction studies of the archaeal virus resolvase SIRV2. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005, 61, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, C.A.; Hynes, R.O. Distribution and evolution of von Willebrand/integrin a domains: Widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell 2002, 13, 3369–3387. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.S.; Houry, W.A. Novel structural and functional insights into the MoxR family of AAA+ ATPases. J. Struct. Biol. 2012, 179, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Snider, J.; Houry, W.A. MoxR AAA+ ATPases: A novel family of molecular chaperones? J. Struct. Biol. 2006, 156, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Scheele, U.; Erdmann, S.; Ungewickell, E.J.; Felisberto-Rodrigues, C.; Ortiz-Lombardia, M.; Garrett, R.A. Chaperone role for proteins p618 and p892 in the extracellular tail development of Acidianus two-tailed virus. J. Virol. 2011, 85, 4812–4821. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Ye, F.; Liew, L.; Liu, D.; Bhushan, S.; Gao, Y.G.; Mueller-Cajar, O. Insights into the mechanism and regulation of the CbbQO-type rubisco activase, a MoxR AAA+ ATPase. Proc. Natl. Acad. Sci. USA 2020, 117, 381–387. [Google Scholar] [CrossRef]
- Wong, K.S.; Bhandari, V.; Janga, S.C.; Houry, W.A. The RavA-ViaA chaperone-like system interacts with and modulates the activity of the fumarate reductase respiratory complex. J. Mol. Biol. 2017, 429, 324–344. [Google Scholar] [CrossRef]
- Krishna, S.S.; Majumdar, I.; Grishin, N.V. Structural classification of zinc fingers: Survey and summary. Nucleic Acids Res. 2003, 31, 532–550. [Google Scholar] [CrossRef]
- To, K.H.; Young, R. Probing the structure of the S105 hole. J. Bacteriol. 2014, 196, 3683–3689. [Google Scholar] [CrossRef]
- Cahill, J.; Young, R. Phage lysis: Multiple genes for multiple barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar]
- Casjens, S.R.; Gilcrease, E.B.; Winn-Stapley, D.A.; Schicklmaier, P.; Schmieger, H.; Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Hatfull, G.F.; Hendrix, R.W. The generalized transducing Salmonella bacteriophage ES18: Complete genome sequence and DNA packaging strategy. J. Bacteriol. 2005, 187, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Desiere, F.; Mahanivong, C.; Hillier, A.J.; Chandry, P.S.; Davidson, B.E.; Brussow, H. Comparative genomics of lactococcal phages: Insight from the complete genome sequence of Lactococcus lactis phage BK5-T. Virology 2001, 283, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Garcia-Heredia, I.; Martin-Cuadrado, A.B.; Ghai, R. Reconstruction of novel cyanobacterial siphovirus genomes from mediterranean metagenomic fosmids. Appl. Environ. Microbiol. 2013, 79, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Millard, A.D.; Pearce, D.; Zwirglmaier, K. Biogeography of bacteriophages at four hydrothermal vent sites in the Antarctic based on g23 sequence diversity. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed]
- DSMZ Webserver (VICTOR). Available online: https://victor.dsmz.de (accessed on 1 June 2020).
- Robinson, C.K.; Wierzchos, J.; Black, C.; Crits-Christoph, A.; Ma, B.; Ravel, J.; Ascaso, C.; Artieda, O.; Valea, S.; Roldan, M.; et al. Microbial diversity and the presence of algae in halite endolithic communities are correlated to atmospheric moisture in the hyper-arid zone of the Atacama desert. Environ. Microbiol. 2015, 17, 299–315. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Liu, Y.; Du, K.; Xu, S.; Wang, Y.; Krupovic, M.; Chen, X. A novel family of tyrosine integrases encoded by the temperate pleolipovirus SNJ2. Nucleic Acids Res. 2018, 46, 2521–2536. [Google Scholar] [CrossRef]
- Gcf_000337035.1 (Hrr. coriense Genome Assembly). Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000337035.1/ (accessed on 1 June 2020).
- Pagaling, E.; Haigh, R.D.; Grant, W.D.; Cowan, D.A.; Jones, B.E.; Ma, Y.; Ventosa, A.; Heaphy, S. Sequence analysis of an archaeal virus isolated from a hypersaline lake in Inner Mongolia, China. BMC Genomics 2007, 8, 410. [Google Scholar] [CrossRef]
- Podell, S.; Ugalde, J.A.; Narasingarao, P.; Banfield, J.F.; Heidelberg, K.B.; Allen, E.E. Assembly-driven community genomics of a hypersaline microbial ecosystem. PLoS ONE 2013, 8, e61692. [Google Scholar] [CrossRef]
- Bath, C.; Cukalac, T.; Porter, K.; Dyall-Smith, M.L. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology 2006, 350, 228–239. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Liu, Y.; Wang, Y.; Zhang, Z.; Oksanen, H.M.; Bamford, D.H.; Chen, X. Identification and characterization of SNJ2, the first temperate pleolipovirus integrating into the genome of the SNJ1-lysogenic archaeal strain. Mol. Microbiol. 2015, 98, 1002–1020. [Google Scholar] [CrossRef]
- Demina, T.A.; Atanasova, N.S.; Pietila, M.K.; Oksanen, H.M.; Bamford, D.H. Vesicle-like virion of Haloarcula hispanica pleomorphic virus 3 preserves high infectivity in saturated salt. Virology 2016, 499, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Dyall-Smith, M.; Tang, S.-L.; Pfeiffer, F. Haloviruses: The bad, the worse and the surprising. Studia Universitatis Babes-Bolyai, Biologia 2019, 64, 51. [Google Scholar]
- Schulze, S.; Adams, Z.; Cerletti, M.; De Castro, R.; Ferreira-Cerca, S.; Fufezan, C.; Gimenez, M.I.; Hippler, M.; Jevtic, Z.; Knuppel, R.; et al. The archaeal proteome project advances knowledge about archaeal cell biology through comprehensive proteomics. Nat. Commun. 2020, 11, 3145. [Google Scholar] [CrossRef] [PubMed]
- Esquivel, R.N.; Schulze, S.; Xu, R.; Hippler, M.; Pohlschroder, M. Identification of Haloferax volcanii pilin N-glycans with diverse roles in pilus biosynthesis, adhesion, and microcolony formation. J. Biol. Chem. 2016, 291, 10602–10614. [Google Scholar] [CrossRef] [PubMed]
- Laass, S.; Monzon, V.A.; Kliemt, J.; Hammelmann, M.; Pfeiffer, F.; Forstner, K.U.; Soppa, J. Characterization of the transcriptome of Haloferax volcanii, grown under four different conditions, with mixed RNA-seq. PLoS ONE 2019, 14, e0215986. [Google Scholar] [CrossRef] [PubMed]
- Kucukyildirim, S.; Behringer, M.; Williams, E.M.; Doak, T.G.; Lynch, M. Estimation of the genome-wide mutation rate and spectrum in the archaeal species Haloferax volcanii. Genetics 2020, 215, 1107–1116. [Google Scholar] [CrossRef]
- Collins, M.; Afolayan, S.; Igiraneza, A.B.; Schiller, H.; Krespan, E.; Beiting, D.P.; Dyall-Smith, M.; Pfeiffer, F.; Pohlschroder, M. Mutations affecting HVO_1357 or HVO_2248 cause hypermotility in Haloferax volcanii, suggesting roles in motility regulation. Genes 2020, 12, 58. [Google Scholar] [CrossRef]
- Dyall-Smith, M.; Palm, P.; Wanner, G.; Witte, A.; Oesterhelt, D.; Pfeiffer, F. Halobacterium salinarum virus ChaoS9, a novel halovirus related to phih1 and phich1. Genes 2019, 10, 194. [Google Scholar] [CrossRef]
- Fullmer, M.S.; Ouellette, M.; Louyakis, A.S.; Papke, R.T.; Gogarten, J.P. The patchy distribution of restriction-modification system genes and the conservation of orphan methyltransferases in halobacteria. Genes 2019, 10, 233. [Google Scholar] [CrossRef]
- Tang, S.L.; Nuttall, S.; Ngui, K.; Fisher, C.; Lopez, P.; Dyall-Smith, M. HF2: A double-stranded DNA tailed haloarchaeal virus with a mosaic genome. Mol. Microbiol. 2002, 44, 283–296. [Google Scholar] [CrossRef]
- Russ, B. Unravelling the transcriptional programme of the haloarchaeal virus HF2; University of Melbourne: Parkville, Victoria, Australia, 2009. [Google Scholar]
- Zecchi, L.; Lo Piano, A.; Suzuki, Y.; Canas, C.; Takeyasu, K.; Ayora, S. Characterization of the Holliday junction resolving enzyme encoded by the Bacillus subtilis bacteriophage spp1. PLoS ONE 2012, 7, e48440. [Google Scholar] [CrossRef] [PubMed]
- Birkenbihl, R.P.; Neef, K.; Prangishvili, D.; Kemper, B. Holliday junction resolving enzymes of archaeal viruses SIRV1 and SIRV2. J. Mol. Biol. 2001, 309, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Subramanian, S. Classification of the treble clef zinc finger: Noteworthy lessons for structure and function evolution. Sci. Rep. 2016, 6, 32070. [Google Scholar] [CrossRef] [PubMed]
- Nagel, C.; Machulla, A.; Zahn, S.; Soppa, J. Several one-domain zinc finger micro-proteins of Haloferax volcanii are important for stress adaptation, biofilm formation, and swarming. Genes 2019, 10, 361. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Host | Sequence Reads 1 | Total Mb | Genome Length (bp) | G + C % | Read Coverage | Accession |
|---|---|---|---|---|---|---|---|
| Hardycor1 | Hrr. coriense | 17,097 | 21.6 | 45,142 | 67.8 | 95× | MT152698 |
| AGCT | CTAG | TGCA | CAGC | CATC | CCAG |
|---|---|---|---|---|---|
| 0 | 0 | 0 | 0.01 | 0.03 | 0.11 |
| First Base | 6-mer Motifs Not Present in Hardycor1 |
|---|---|
| A | ACATGT, AGATCT, AGCGCT, AGTACT, ATTAAT |
| C | CACGTG, CCATGG, CTCGAG, CTRYAG |
| G | GAATTC, GACGTC, GATATC, GCATGC, GGCGCC, GGTACC, GGGCCC, GRGCYC, GTATAC |
| T | TGGCCA, TGTACA, TTCGAA, TTTAAA |
| Hardycor1 Region (nt); Length | Hardycor1 Locus_Tag (Gene) | Matching Sequence (Accession) | Matched Region (nt); Name/Gene | Target Locus_Tag (Accession) | % Identity (E-Value) |
|---|---|---|---|---|---|
| 18158–18692; 540 bp | hrrhc1_120 (tmp) | Halorubrum sp. RHB-C (CP053941.1) | 2930116–2930650; tape measure protein | HPS36_14875 (QKG94091.1) | 66% (7 × 10−28) |
| 25604–26035; 436 bp | hrrhc1_160 (Hyp) | Halovirus HCTV-1 (KC292029.1) | 43575–43153; hypothetical protein | DNAM5_77 (AGM11938.1) | 69% (2 × 10−21) |
| Start | Stop | Locus Tag | Length | Direction | Gene | Product | Protein Homologs a |
|---|---|---|---|---|---|---|---|
| 138 | 791 | HrrHc1_005 | 654 | + | hypothetical protein | E3374_RS16605 [Halorhabdus sp. H27] | |
| 788 | 1012 | HrrHc1_010 | 225 | + | CxxC motif protein | ||
| 1009 | 1323 | HrrHc1_015 | 315 | + | hypothetical protein | ||
| 1320 | 1883 | HrrHc1_020 | 564 | + | dam | probable Dam methylase | DJ70_12660 [Halorubrum halodurans] |
| 1880 | 2260 | HrrHc1_025 | 381 | + | CxxC motif protein | ||
| 2337 | 3560 | HrrHc1_030 | 1224 | + | terL | large subunit terminase TerL | L593_06050 [Salinarchaeum sp. Harcht-Bsk1] |
| 3809 | 5407 | HrrHc1_035 | 1599 | + | por | portal protein Por | FE783_12715 [Paenibacillus mesophilus] |
| 5412 | 7382 | HrrHc1_040 | 1971 | + | muf | SPP1 gp7 family protein MuF | CMK96_05475 [Pseudomonas sp.] |
| 7479 | 8501 | HrrHc1_045 | 1023 | + | hypothetical protein | ||
| 8506 | 8820 | HrrHc1_050 | 315 | – | CxxC motif protein | ||
| 8986 | 10,470 | HrrHc1_055 | 1485 | + | hypothetical protein | Natgr_1848 [Natronobacterium gregoryi SP2] | |
| 10,474 | 10,911 | HrrHc1_060 | 438 | + | hypothetical protein | ||
| 10,913 | 11,989 | HrrHc1_065 | 1077 | + | mcp | major capsid protein Mcp | IEX84_RS06545 [Halarchaeum rubridurum] |
| 12,070 | 12,501 | HrrHc1_070 | 432 | + | hypothetical protein | ||
| 12,505 | 12,933 | HrrHc1_075 | 429 | + | DUF1073 domain protein | ||
| 12,935 | 13,288 | HrrHc1_080 | 354 | + | nep1 | neck protein Nep1 | G9C82_17265 [Haloarcula sp. R1-2] |
| 13,285 | 13,710 | HrrHc1_085 | 426 | + | hypothetical protein | ||
| 13,707 | 14,201 | HrrHc1_090 | 495 | + | hypothetical protein | ||
| 14,337 | 15,266 | HrrHc1_095 | 930 | – | hypothetical protein | ||
| 15,676 | 15,879 | HrrHc1_100 | 204 | + | hypothetical protein | ||
| 15,883 | 17,115 | HrrHc1_105 | 1233 | + | hypothetical protein | AArcSl_1282 [Halalkaliarchaeum desulfuricum] | |
| 17,143 | 17,613 | HrrHc1_110 | 471 | + | hypothetical protein | G6M89_09280 [Natronolimnobius sp. AArcel1] | |
| 17,688 | 17,894 | HrrHc1_115 | 207 | + | hypothetical protein | ||
| 17,894 | 20,005 | HrrHc1_120 | 2112 | + | tmp | tape measure protein Tmp | C484_10631 [Natrialba taiwanensis] |
| 20,007 | 20,552 | HrrHc1_125 | 546 | + | hypothetical protein | ||
| 20,554 | 21,759 | HrrHc1_130 | 1206 | + | hypothetical protein | BBD46_16545 [Natrialba sp. SSL1] | |
| 21,756 | 22,067 | HrrHc1_135 | 312 | + | hypothetical protein | ||
| 22,069 | 22,527 | HrrHc1_140 | 459 | + | hypothetical protein | ||
| 22,599 | 24,518 | HrrHc1_145 | 1920 | + | hypothetical protein | GS429_08425 [Natronorubrum sp. JWXQ-INN-674] | |
| 24,574 | 24,813 | HrrHc1_150 | 240 | + | hypothetical protein | ||
| 24,825 | 25,175 | HrrHc1_155 | 351 | + | predicted membrane protein | ||
| 25,290 | 26,255 | HrrHc1_160 | 966 | + | hypothetical protein | DNAM5_77 [HCTV-1], HHTV2_37 [HHTV-2] | |
| 26,324 | 26,917 | HrrHc1_165 | 594 | + | hypothetical protein | EPY72_RS18050 [Halorussus sp. LYG-36] | |
| 27,104 | 27,502 | HrrHc1_170 | 399 | – | CxxC motif protein | ||
| 27,499 | 27,936 | HrrHc1_175 | 438 | – | hjc | H-J resolvase b Hjc | BRC93_05600 [Halobacteriales archaeon] |
| 28,034 | 28,390 | HrrHc1_180 | 357 | – | hypothetical protein | ||
| 28,390 | 28,674 | HrrHc1_185 | 285 | – | hypothetical protein | ||
| 28,671 | 29,117 | HrrHc1_190 | 447 | – | hypothetical protein | HHTV1_58 [HHTV-1] | |
| 29,114 | 29,314 | HrrHc1_195 | 201 | – | CxxC motif protein | ||
| 29,311 | 29,610 | HrrHc1_200 | 300 | – | CxxC motif protein | ||
| 29,607 | 29,867 | HrrHc1_205 | 261 | – | CxxC motif protein | ||
| 29,860 | 31,395 | HrrHc1_210 | 1536 | – | nucleic acid binding domain protein | HCTV2_73 [HCTV-2] | |
| 31,392 | 32,585 | HrrHc1_215 | 1194 | – | hypothetical protein | HCTV2_75 [HCTV-2], HHTV2_88 [HHTV-2] | |
| 32,774 | 32,938 | HrrHc1_220 | 165 | – | CxxC motif protein | ||
| 32,935 | 33,327 | HrrHc1_225 | 393 | – | CxxC motif protein | ||
| 33,324 | 35,321 | HrrHc1_230 | 1998 | – | hypothetical protein | DM826_07300 [Halonotius sp. F13-13] | |
| 35,523 | 37,079 | HrrHc1_235 | 1557 | – | CxxC motif protein | ||
| 37,288 | 38,694 | HrrHc1_240 | 1407 | – | aaa | AAA ATPase | HHTV2_10 [HHTV-2], HCTV2_83 [HCTV-2] |
| 38,836 | 40,968 | HrrHc1_245 | 2133 | – | vwa | vWA and MIDAS domain protein | HCTV2_79 [HCTV-2], HHTV2_3 [HHTV-2] |
| 41,082 | 42,461 | HrrHc1_250 | 1380 | – | hypothetical protein | ||
| 42,458 | 43,084 | HrrHc1_255 | 627 | – | hypothetical protein | ||
| 43,162 | 43,923 | HrrHc1_260 | 762 | + | hypothetical protein | ||
| 44,232 | 44,936 | HrrHc1_265 | 705 | + | hypothetical protein |
| Provirus | Length (nt) | Archival Virus Stock b | G + C% | Read Coverage | Assembled Contig | Affiliation (Accession) | Comments |
|---|---|---|---|---|---|---|---|
| Humcor1 | 46,474 | CC1 | 62.5 | 184 | circular dsDNA | siphovirus (MW344765) | Matches Hrr. coriense Ch2T (nt 170617–217091; AOJL01000026). |
| Humcor2 | 11,758 | HC1 | 62.5 | 54 | circular dsDNA | pleolipovirus (MW344764) | Matches Hrr. coriense Ch2T (nt 11011–23038; AOJL01000020). |
| Halfgib1 | 16,280 | HG1 | 56.5 | 470 | circular dsDNA | pleolipovirus (MW344766) | Matches Hfx. gibbonsii Ma2.38T (nt 269,983–286,444; AOLJ01000022). |
| Harhisp1 | 19,481 | HH1 | 53.2 | 1403 | circular dsDNA | pleolipovirus (MW344767) | Matches Har. hispanica Y27T (nt 2722239 -2741719; CP006884) |
| Halfvol1 | 20,573 | HV2 | 57.6 | 77 | circular dsDNA | pleolipovirus | Matches Hfx. volcanii DS2T (nt 231453–252025; CP001956) |
| Halfvol2 | 12,275 | HV2 | 62.2 | 165 | circular dsDNA | novel group | Matches Hfx. volcanii DS2T (nt 329565–341853; CP001956) |
| Halfvol3 | 12,527 | - | 59.3 | - | circular dsDNA | pleolipovirus | Matches Hfx. volcanii DS2T (nt 1307486–1294960) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyall-Smith, M.; Pfeiffer, F.; Chiang, P.-W.; Tang, S.-L. The Novel Halovirus Hardycor1, and the Presence of Active (Induced) Proviruses in Four Haloarchaea. Genes 2021, 12, 149. https://doi.org/10.3390/genes12020149
Dyall-Smith M, Pfeiffer F, Chiang P-W, Tang S-L. The Novel Halovirus Hardycor1, and the Presence of Active (Induced) Proviruses in Four Haloarchaea. Genes. 2021; 12(2):149. https://doi.org/10.3390/genes12020149
Chicago/Turabian StyleDyall-Smith, Mike, Friedhelm Pfeiffer, Pei-Wen Chiang, and Sen-Lin Tang. 2021. "The Novel Halovirus Hardycor1, and the Presence of Active (Induced) Proviruses in Four Haloarchaea" Genes 12, no. 2: 149. https://doi.org/10.3390/genes12020149
APA StyleDyall-Smith, M., Pfeiffer, F., Chiang, P.-W., & Tang, S.-L. (2021). The Novel Halovirus Hardycor1, and the Presence of Active (Induced) Proviruses in Four Haloarchaea. Genes, 12(2), 149. https://doi.org/10.3390/genes12020149