
Genomics Reveal Admixture and Unexpected Patterns of Diversity in a Parapatric Pair of Butterflies

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Data
2.2. Genomic Assembly and Bioinformatics
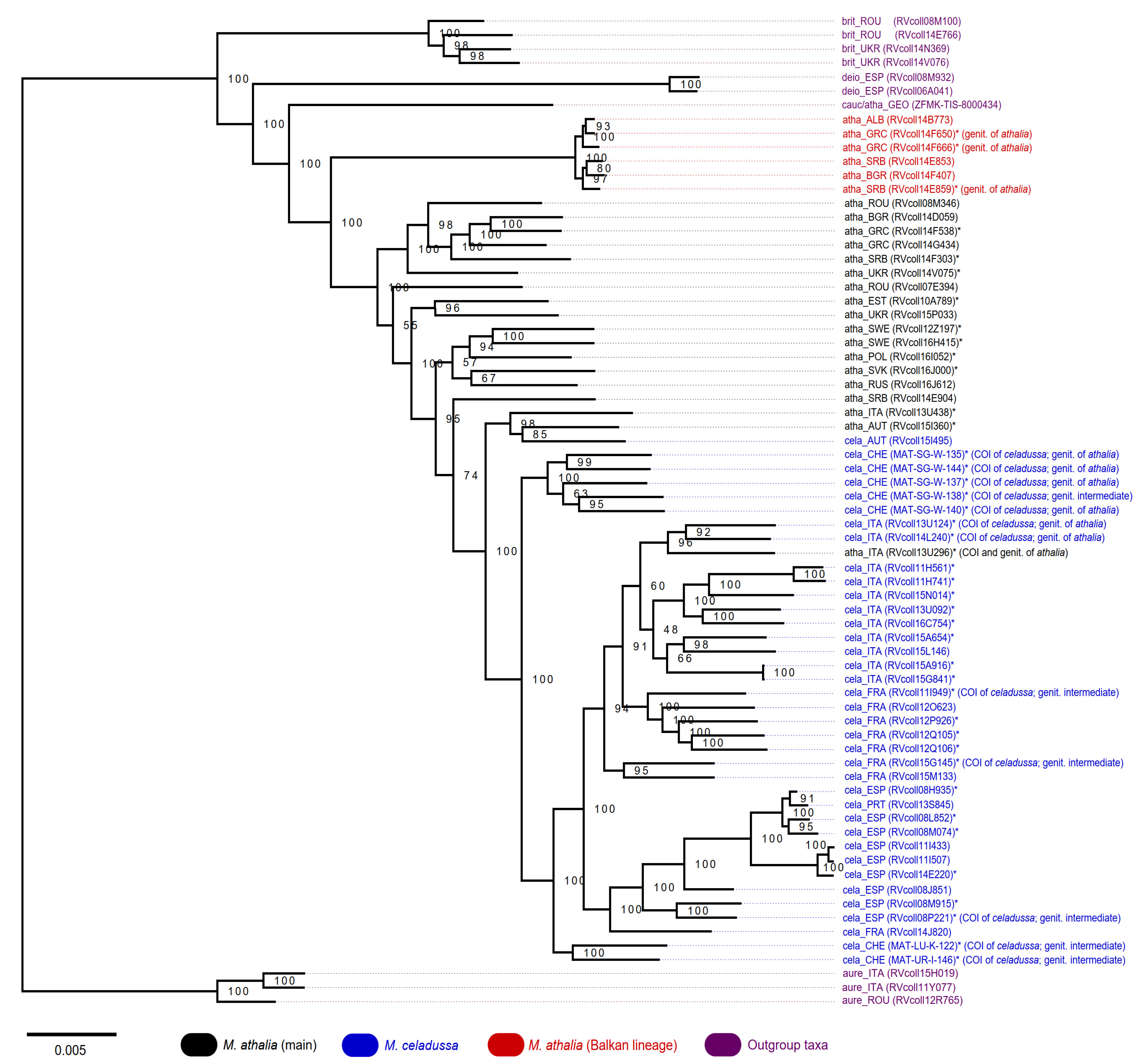
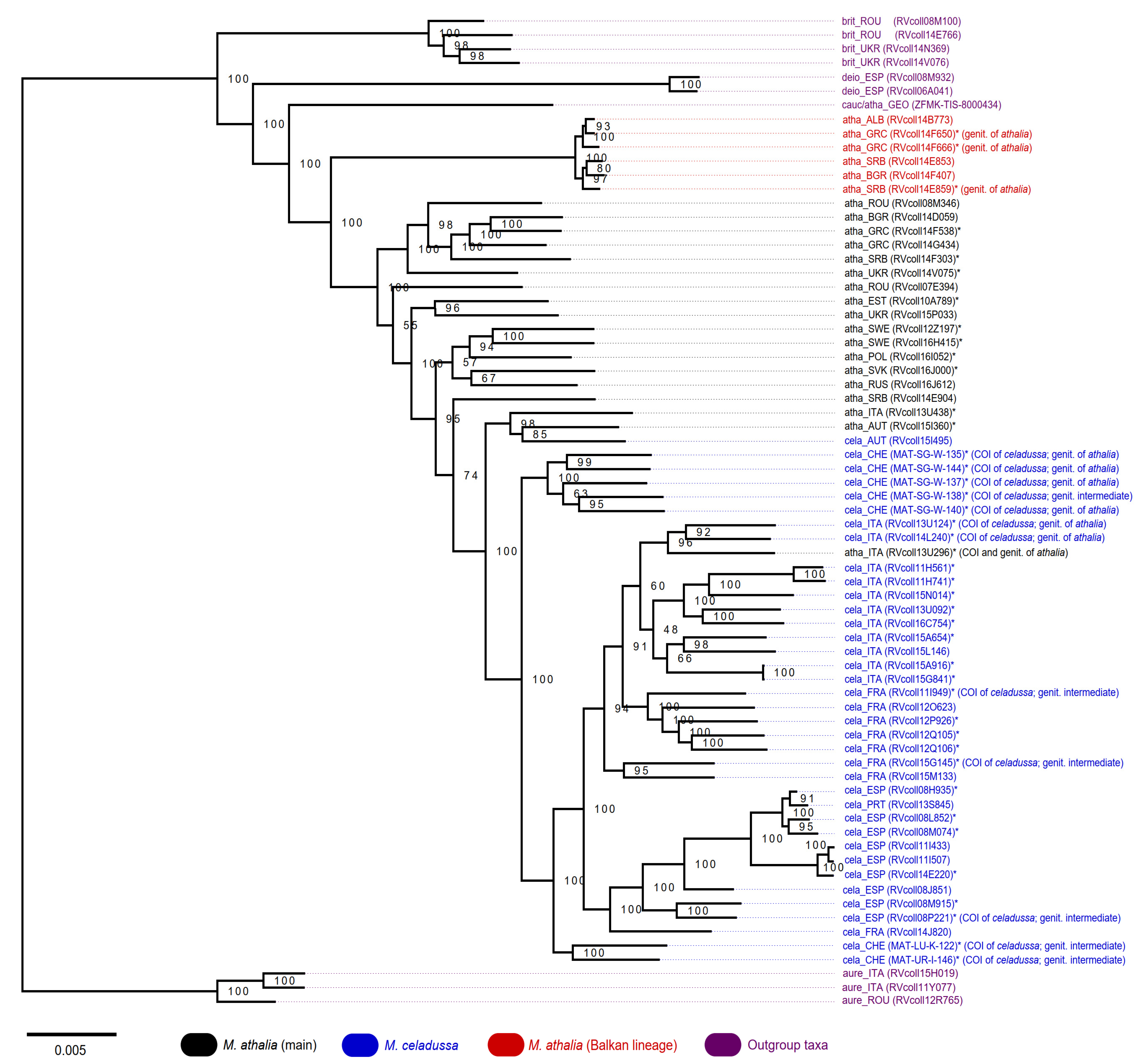
2.3. Phylogenetic Analysis
2.4. Analyses of Genomic Diversity and Admixture
2.5. Population History
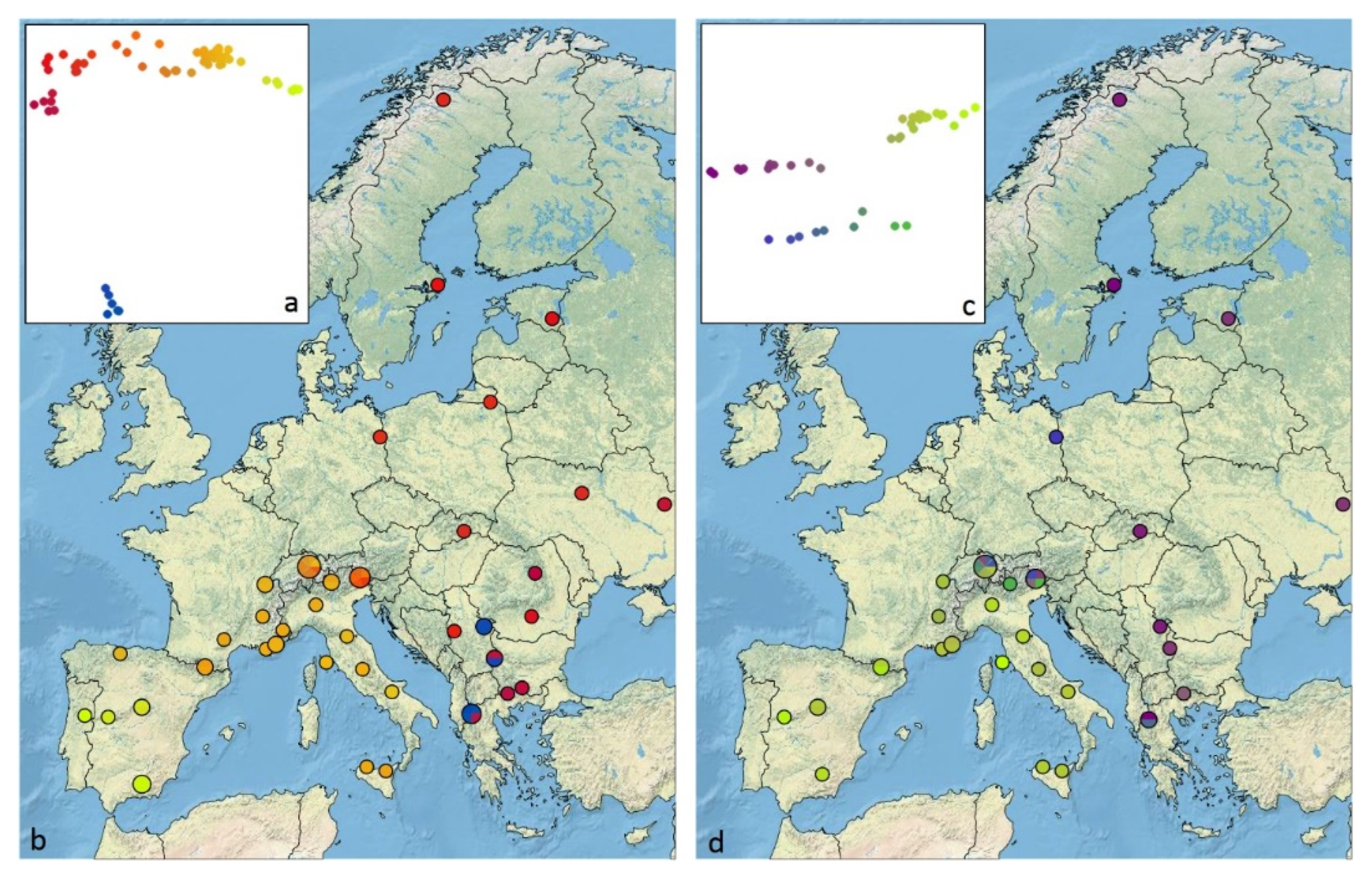
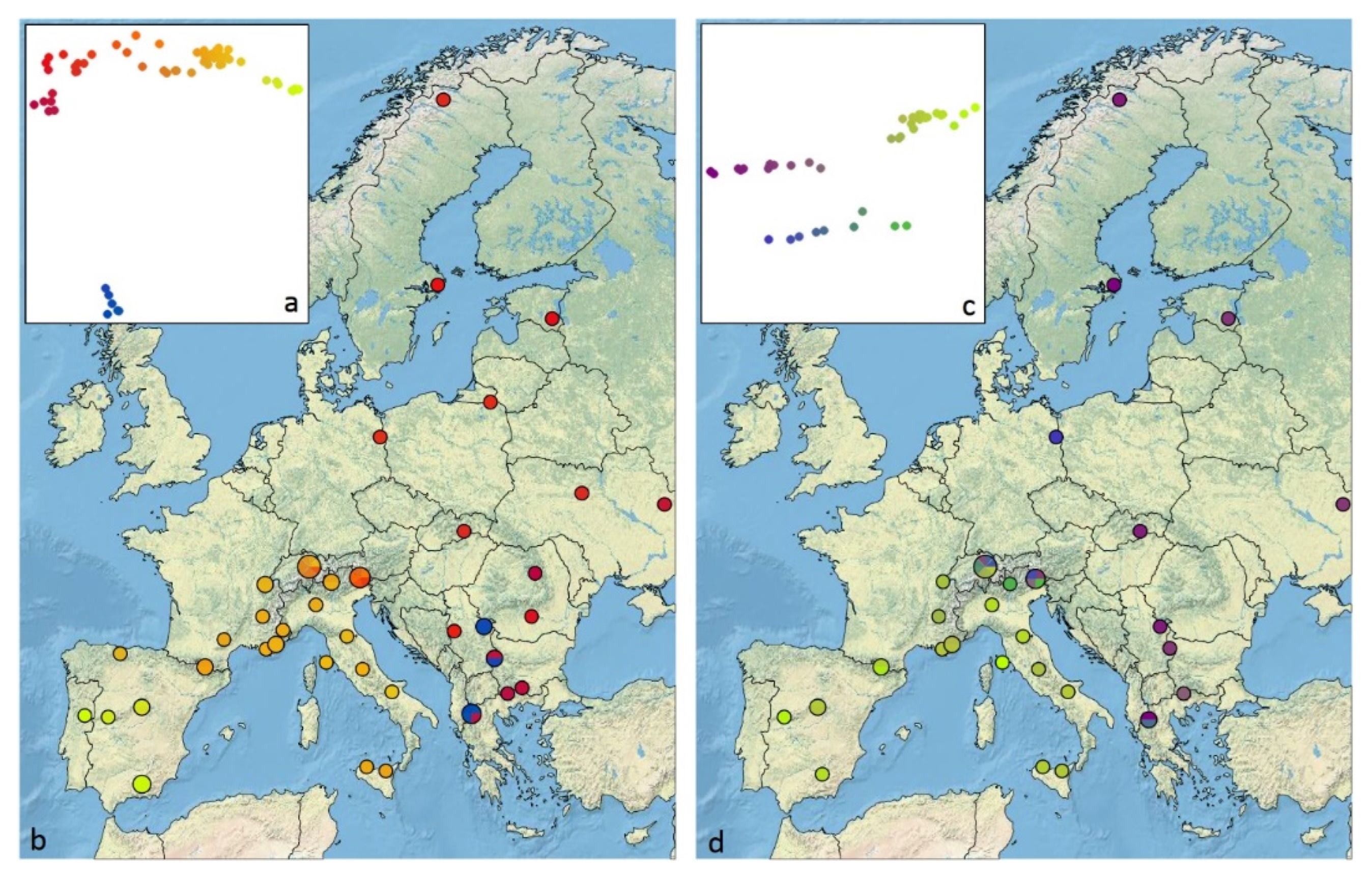
2.6. Isolation by Distance and Geographic Representation of Genetic Differentiation
3. Results
3.1. Patterns of COI Variability
3.2. Morphometrics
3.3. Overview of ddRAD Data
3.4. ddRAD Phylogeny
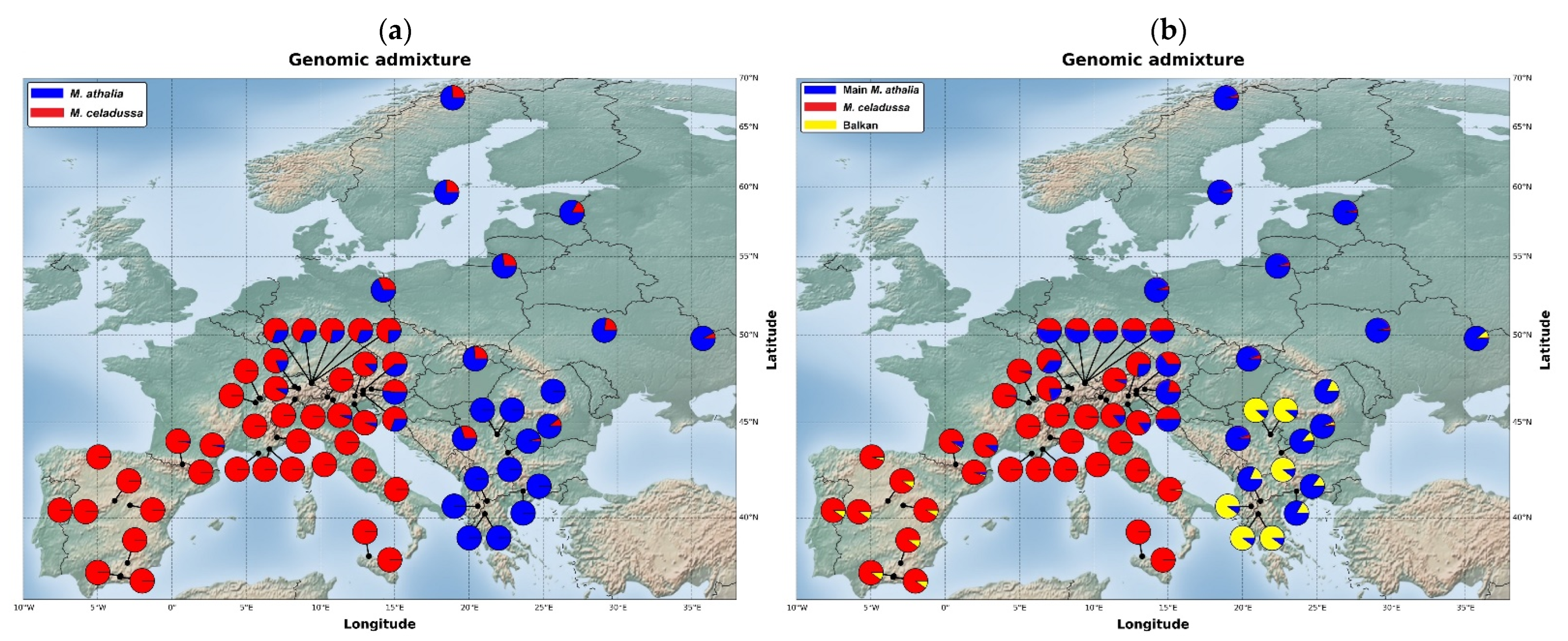
3.5. Genomic Diversity and Admixture
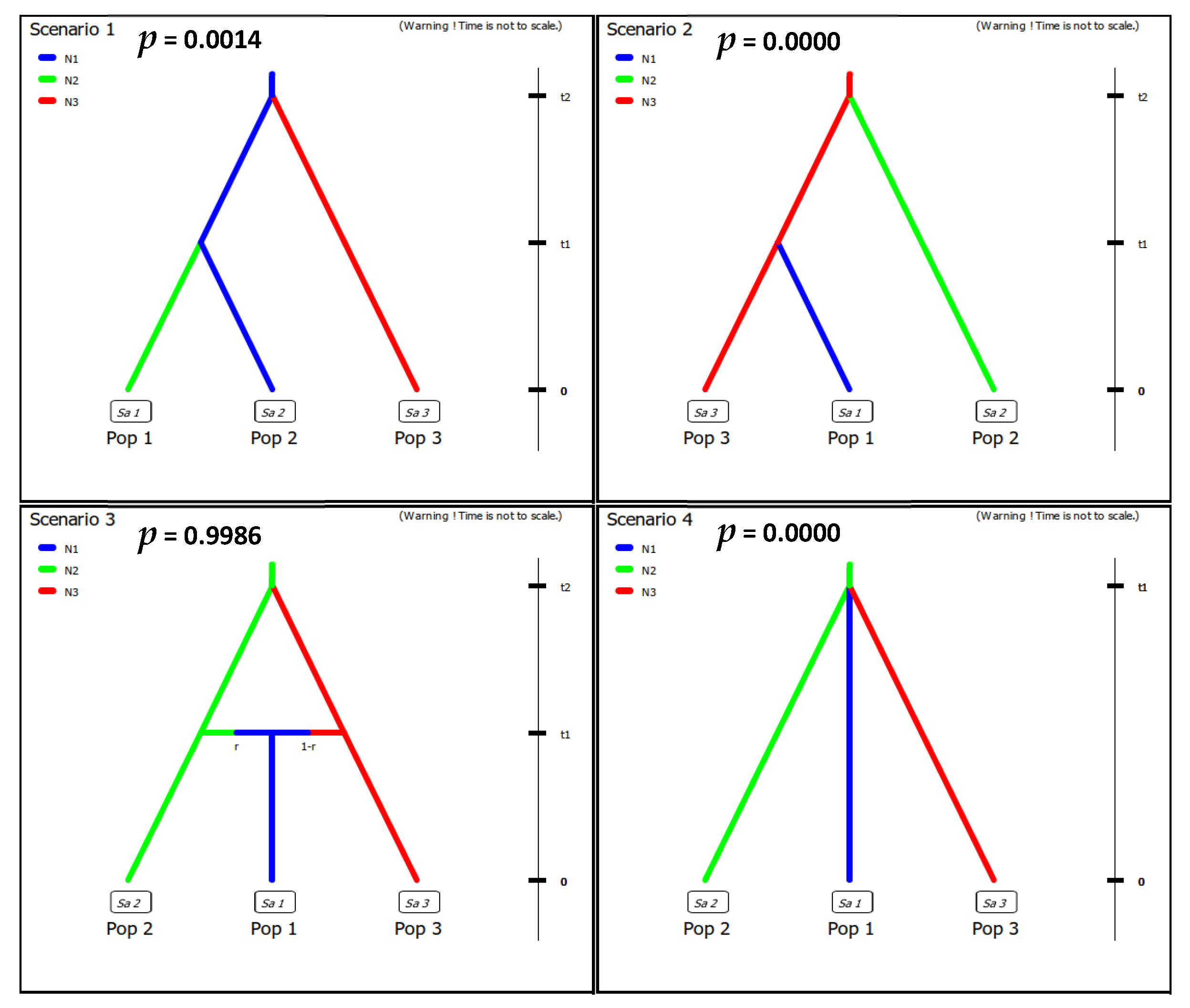
3.6. Population History
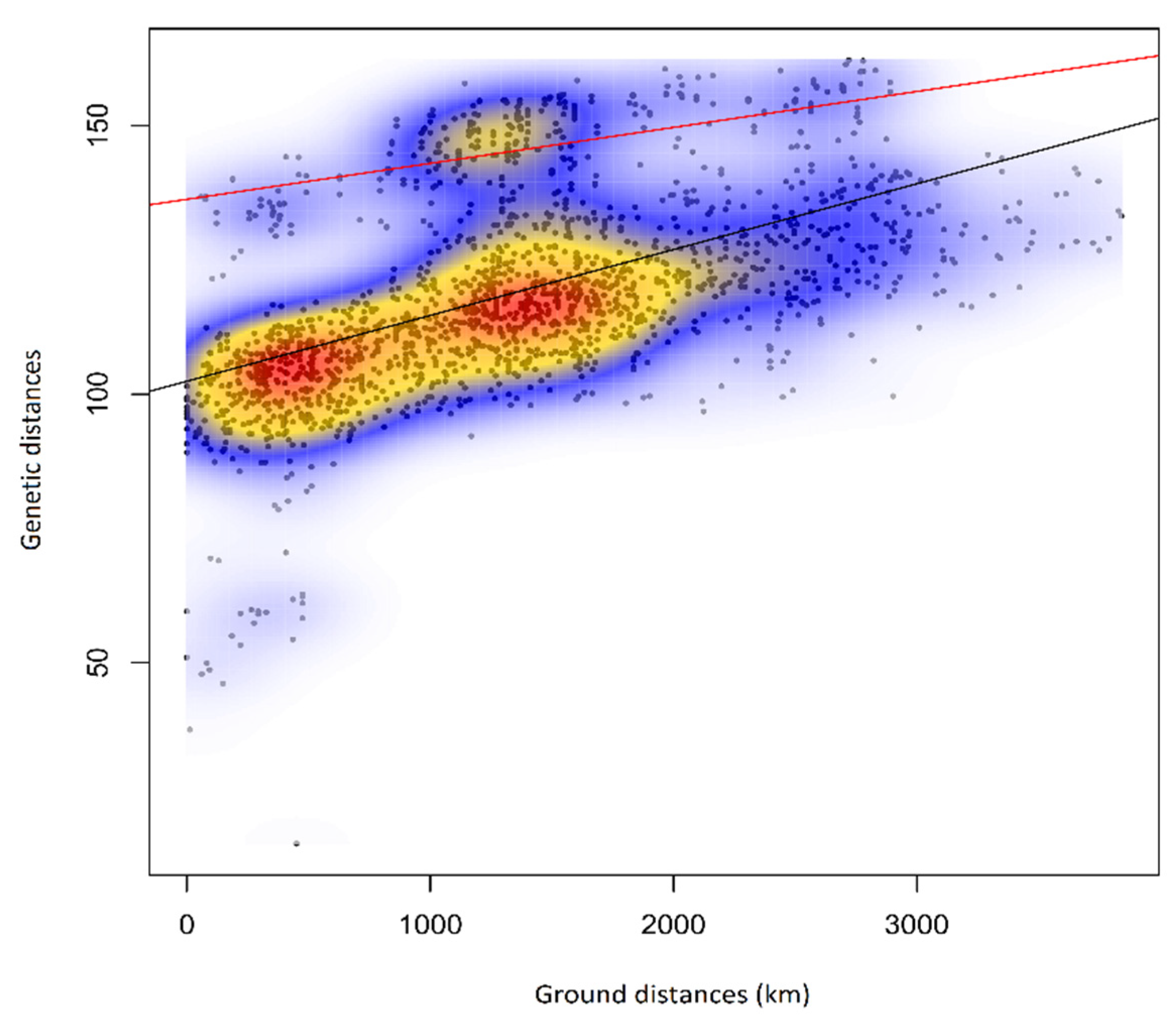
3.7. Isolation by Distance
4. Discussion
4.1. Genetic Structure and Levels of Admixture in M. athalia and M. celadussa
4.2. Patterns of Variability in Male Genital Morphology
4.3. The Evolutionary Origin of the M. athalia–M. celadussa Distribution
4.4. Species Delimitation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sei, M.; Porter, A.H. Delimiting species boundaries and the conservation genetics of the endangered maritime ringlet butterfly (Coenonympha nipisiquit McDunnough). Mol. Ecol. 2007, 16, 3313–3325. [Google Scholar] [CrossRef]
- Abbott, R.; Albach, D.; Ansell, S.; Arntzen, J.W.; Baird, S.J.E.; Bierne, N.; Boughman, J.; Brelsford, A.; Buerkle, C.A.; Buggs, R.; et al. Hybridization and speciation. J. Evol. Biol. 2013, 26, 229–246. [Google Scholar] [CrossRef] [Green Version]
- Fišer, C.; Robinson, C.T.; Malard, F. Cryptic species as a window into the paradigm shift of the species concept. Mol. Ecol. 2018, 27, 613–635. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, K. Species Concepts and Species Delimitation. Syst. Biol. 2007, 56, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Eberhard, W.G. Sexual Selection and Animal Genitalia; Harvard University Press: Cambridge, MA, USA, 1985; Volume 244. [Google Scholar]
- Mikkola, K. The lock-and-key mechanisms in the internal genitalia of the noctuid and geometrid moths (Lepidoptera) in relation to the speciation concepts. Folia Baeriana 1993, 6, 149–157. [Google Scholar]
- Ennos, R.A.; French, G.C.; Hollingsworth, P.M. Conserving taxonomic complexity. Trends Ecol. Evol. 2005, 20, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, Q.D.; Raven, P.H.; Wilson, E.O. Taxonomy: Impediment or Expedient? Science 2004, 303, 285. [Google Scholar] [CrossRef] [Green Version]
- Sites, J.W.; Marshall, J.C. Delimiting species: A Renaissance issue in systematic biology. Trends Ecol. Evol. 2003, 18, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Mutanen, M.; Kaitala, A.; Mönkkönen, M. Genital variation within and between three closely related Euxoa moth species: Testing the lock-and-key hypothesis. J. Zool. 2006, 268, 109–119. [Google Scholar] [CrossRef]
- Flantua, S.G.A.; Payne, D.; Borregaard, M.K.; Beierkuhnlein, C.; Steinbauer, M.J.; Dullinger, S.; Essl, F.; Irl, S.D.H.; Kienle, D.; Kreft, H. Snapshot isolation and isolation history challenge the analogy between mountains and islands used to understand endemism. Glob. Ecol. Biogeogr. 2020, 29, 1651–1673. [Google Scholar] [CrossRef]
- Knief, U.; Bossu, C.M.; Saino, N.; Hansson, B.; Poelstra, J.; Vijay, N.; Weissensteiner, M.; Wolf, J.B.W. Epistatic mutations under divergent selection govern phenotypic variation in the crow hybrid zone. Nat. Ecol. Evol. 2019, 3, 570–576. [Google Scholar] [CrossRef]
- Leaché, A.D.; Fujita, M.K.; Minin, V.N.; Bouckaert, R.R. Species delimitation using genome-wide SNP Data. Syst. Biol. 2014, 63, 534–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberle, J.; Ahrens, D.; Mayer, C.; Niehuis, O.; Misof, B. A plea for standardized nuclear markers in metazoan DNA taxonomy. Trends Ecol. Evol. 2020, 35, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.G. A descriptive catalogue of the genus Mellicta Billberg (Lepidoptera: Nymphalidae) and its species, with supplementary notes on the genera Melitaea and Euphydryas. Trans. R. Entomol. Soc. Lond. 1955, 106, 1–127. [Google Scholar] [CrossRef]
- Van Oorschot, H.; Coutsis, J.G. The Genus Melitaea Fabricius, 1807: Taxonomy and Systematics with Special Reference to the Male Genitalia:(Lepidoptera, Nymphalidae, Nymphalinae); Tshikolovets Publications: Pardubice, Czech Republic, 2014; ISBN 8090490042. [Google Scholar]
- Platania, L.; Menchetti, M.; Dincă, V.; Corbella, C.; Kay-Lavelle, I.; Vila, R.; Wiemers, M.; Schweiger, O.; Dapporto, L. Assigning occurrence data to cryptic taxa improves climatic niche assessments: Biodecrypt, a new tool tested on European butterflies. Glob. Ecol. Biogeogr. 2020, 29, 1852–1865. [Google Scholar] [CrossRef]
- Dincă, V.; Dapporto, L.; Somervuo, P.; Vodă, R.; Cuvelier, S.; Gascoigne-Pees, M.; Huemer, P.; Mutanen, M.; Hebert, P.D.N.; Vila, R. High resolution DNA barcode library for European butterflies reveals continental patterns of mitochondrial genetic diversity. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.G. Classification of European Butterflies; Collins: New York, NY, USA, 1975; ISBN 0002196247. [Google Scholar]
- Leneveu, J.; Chichvarkhin, A.; Wahlberg, N. Varying rates of diversification in the genus Melitaea (Lepidoptera: Nymphalidae) during the past 20 million years. Biol. J. Linn. Soc. 2009, 97, 346–361. [Google Scholar] [CrossRef] [Green Version]
- Dincă, V.; Montagud, S.; Talavera, G.; Hernández-Roldán, J.; Munguira, M.L.; García-Barros, E.; Hebert, P.D.N.; Vila, R. DNA barcode reference library for Iberian butterflies enables a continental-scale preview of potential cryptic diversity. Sci. Rep. 2015, 5, 12395. [Google Scholar] [CrossRef] [PubMed]
- Wiemers, M.; Balletto, E.; Dincă, V.; Fric, Z.F.; Lamas, G.; Lukhtanov, V.; Munguira, M.L.; van Swaay, C.A.M.; Vila, R.; Vliegenthart, A. An updated checklist of the European butterflies (Lepidoptera, Papilionoidea). Zookeys 2018, 811, 9–45. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, D.A.R.; Overcast, I. ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 2019, 36, 2592–2594. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Estoup, A.; Cornuet, J.-M.; Dehne-Garcia, A.; Loire, E. New Version of the Computer Program DIYABC (DIYABC v2. 1.0): A User-Friendly Approach to Approximate Bayesian Computation for Inference on Population History Using Molecular Markers; ResearchGate: Berlin, Germany, 2015. [Google Scholar]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Van Etten, J. R Package gdistance: Distances and Routes on Geographical Grids. J. Stat. Softw. 2017, 76, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Shafer, A.B.A.; Peart, C.R.; Tusso, S.; Maayan, I.; Brelsford, A.; Wheat, C.W.; Wolf, J.B.W. Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol. Evol. 2017, 8, 907–917. [Google Scholar] [CrossRef]
- Huemer, P.; Wiesmair, B. DNA-Barcoding der Tagfalter (Lepidoptera, Papilionoidea) Österreichs. Unbekannte genetische Vielfalt im Zentrum Österreichs. Wiss. J. Tirol. Landesmuseen 2017, 10, 8–33. [Google Scholar]
- Litman, J.; Chittaro, Y.; Birrer, S.; Praz, C.; Wermeille, E.; Fluri, M.; Stalling, T.; Schmid, S.; Wyler, S.; Gonseth, Y. A DNA barcode reference library for Swiss butterflies and forester moths as a tool for species identification, systematics and conservation. PLoS ONE 2018, 13, e0208639. [Google Scholar] [CrossRef]
- Achtelik, G. Molekularbiologische Analyse der genetischen Diversität des Melitaea Athalia/Celadussa-Komplexes (Lepidoptera: Nymphalidae) unter Anwendung der ISSR-PCR auf Art-, Unterart-und Populationsebene. Ph.D. Thesis, Ruhr-Universität, Bochum, Germany, 2006. [Google Scholar]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.; Cosson, J. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef]
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef]
- Toews, D.P.L.; Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 2012, 21, 3907–3930. [Google Scholar] [CrossRef]
- Dapporto, L.; Vodă, R.; Dincă, V.; Vila, R. Comparing population patterns for genetic and morphological markers with uneven sample sizes. An example for the butterfly Maniola jurtina. Methods Ecol. Evol. 2014, 5, 834–843. [Google Scholar] [CrossRef]
- Rosser, N.; Shirai, L.T.; Dasmahapatra, K.K.; Mallet, J.; Freitas, A.V.L. The Amazon river is a suture zone for a polyphyletic group of co-mimetic heliconiine butterflies. Ecography 2021, 44, 177–187. [Google Scholar] [CrossRef]
- De La Torre, A.; Ingvarsson, P.K.; Aitken, S.N. Genetic architecture and genomic patterns of gene flow between hybridizing species of Picea. Heredity 2015, 115, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Mavárez, J.; Salazar, C.A.; Bermingham, E.; Salcedo, C.; Jiggins, C.D.; Linares, M. Speciation by hybridization in Heliconius butterflies. Nature 2006, 441, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Salazar, C.; Jiggins, C.D.; Taylor, J.E.; Kronforst, M.R.; Linares, M. Gene flow and the genealogical history of Heliconius heurippa. BMC Evol. Biol. 2008, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, V.L.; Dimond, L.; Harrison, J.S.; Velázquez, C.C.S.; Zieba, J.T.; Burton, R.S.; Edmands, S. Interpopulation hybridization results in widespread viability selection across the genome in Tigriopus californicus. BMC Genet. 2011, 12, 54. [Google Scholar] [CrossRef] [Green Version]
- Nadeau, N.J.; Salazar, P.; Morrison, A.; Jiggins, C.D.; Ruiz, M.; Papa, R.; Counterman, B.; Medina, J.A.; Ortiz-Zuazaga, H.; McMillan, W.O. Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato. Genome Res. 2014, 24, 1316–1333. [Google Scholar] [CrossRef] [Green Version]
- Dincă, V.; Lee, K.M.; Vila, R.; Mutanen, M. The conundrum of species delimitation: A genomic perspective on a mitogenetically super-variable butterfly. Proc. R. Soc. B 2019, 286, 20191311. [Google Scholar] [CrossRef] [PubMed]
- Koren, T.; Beretta, S.; Črne, M.; Verovnik, R. On the distribution of Pyrgus malvoides (Elwes & Edwards, 1897)(Lepidoptera: Hesperiidae) at the eastern part of its range. Entomol. Gaz. 2013, 64, 225–234. [Google Scholar]
- Lin, D.; Bi, K.; Conroy, C.J.; Lacey, E.A.; Schraiber, J.G.; Bowie, R.C.K. Mito-nuclear discordance across a recent contact zone for California voles. Ecol. Evol. 2018, 8, 6226–6241. [Google Scholar] [CrossRef] [PubMed]
- Larmuseau, M.H.D.; Raeymaekers, J.A.M.; Hellemans, B.; Van Houdt, J.K.J.; Volckaert, F.A.M. Mito-nuclear discordance in the degree of population differentiation in a marine goby. Heredity 2010, 105, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, J.C.; Sites, J.W., Jr. A comparison of nuclear and mitochondrial cline shapes in a hybrid zone in the Sceloporus grammicus complex (Squamata; Phrynosomatidae). Mol. Ecol. 2001, 10, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.; Lee, K.M.; Mutanen, M. Mitonuclear discordance in wolf spiders: Genomic evidence for species integrity and introgression. Mol. Ecol. 2018, 27, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Mullen, S.P.; Dopman, E.B.; Harrison, R.G. Hybrid zone origins, species boundaries, and the evolution of wing-pattern diversity in a polytypic species complex of North American admiral butterflies (Nymphalidae: Limenitis). Evolution 2008, 62, 1400–1417. [Google Scholar] [CrossRef] [PubMed]
- Gavrilets, S.; Li, H.; Vose, M.D. Patterns of parapatric speciation. Evolution 2000, 54, 1126–1134. [Google Scholar] [CrossRef]
- Lukhtanov, V.A.; Kandul, N.P.; Plotkin, J.B.; Dantchenko, A.V.; Haig, D.; Pierce, N.E. Reinforcement of pre-zygotic isolation and karyotype evolution in Agrodiaetus butterflies. Nature 2005, 436, 385–389. [Google Scholar] [CrossRef]
- Barton, N.H.; Turelli, M. Spatial waves of advance with bistable dynamics: Cytoplasmic and genetic analogues of Allee effects. Am. Nat. 2011, 178, E48–E75. [Google Scholar] [CrossRef] [Green Version]
- Dincă, V.; Wiklund, C.; Lukhtanov, V.A.; Kodandaramaiah, U.; Norén, K.; Dapporto, L.; Wahlberg, N.; Vila, R.; Friberg, M. Reproductive isolation and patterns of genetic differentiation in a cryptic butterfly species complex. J. Evol. Biol. 2013, 26, 2095–2106. [Google Scholar] [CrossRef] [Green Version]
- Habel, J.C.; Vila, R.; Vodă, R.; Husemann, M.; Schmitt, T.; Dapporto, L. Differentiation in the marbled white butterfly species complex driven by multiple evolutionary forces. J. Biogeogr. 2017, 44, 433–445. [Google Scholar] [CrossRef]
- Platania, L.; Vodă, R.; Dincă, V.; Talavera, G.; Vila, R.; Dapporto, L. Integrative analyses on Western Palearctic Lasiommata reveal a mosaic of nascent butterfly species. J. Zool. Syst. Evol. Res. 2020, 58, 809–822. [Google Scholar] [CrossRef]
- Pigot, A.L.; Tobias, J.A. Species interactions constrain geographic range expansion over evolutionary time. Ecol. Lett. 2013, 16, 330–338. [Google Scholar] [CrossRef]
- Waters, J.M.; Fraser, C.I.; Hewitt, G.M. Founder takes all: Density-dependent processes structure biodiversity. Trends Ecol. Evol. 2013, 28, 78–85. [Google Scholar] [CrossRef]
- Vodă, R.; Dapporto, L.; Dincă, V.; Vila, R. Why do cryptic species tend not to co-occur? A case study on two cryptic pairs of butterflies. PLoS ONE 2015, 10, e0117802. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, T.; Louy, D.; Zimmermann, E.; Habel, J.C. Species radiation in the Alps: Multiple range shifts caused diversification in Ringlet butterflies in the European high mountains. Org. Divers. Evol. 2016, 16, 791–808. [Google Scholar] [CrossRef]
- Dincă, V.; Zakharov, E.V.; Hebert, P.D.N.; Vila, R. Complete DNA barcode reference library for a country’s butterfly fauna reveals high performance for temperate Europe. Proc. R. Soc. B Biol. Sci. 2011, 278, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratton, P.; Trucchi, E.; Trasatti, A.; Riccarducci, G.; Marta, S.; Allegrucci, G.; Cesaroni, D.; Sbordoni, V. Testing classical species properties with contemporary data: How “bad species” in the brassy ringlets (Erebia tyndarus complex, Lepidoptera) turned good. Syst. Biol. 2016, 65, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Zinetti, F.; Dapporto, L.; Vovlas, A.; Chelazzi, G.; Bonelli, S.; Balletto, E.; Ciofi, C. When the rule becomes the exception. No evidence of gene flow between two Zerynthia cryptic butterflies suggests the emergence of a new model group. PLoS ONE 2013, 8, e65746. [Google Scholar] [CrossRef] [Green Version]
- Daïnou, K.; Blanc-Jolivet, C.; Degen, B.; Kimani, P.; Ndiade-Bourobou, D.; Donkpegan, A.S.L.; Tosso, F.; Kaymak, E.; Bourland, N.; Doucet, J.-L. Revealing hidden species diversity in closely related species using nuclear SNPs, SSRs and DNA sequences—A case study in the tree genus Milicia. BMC Evol. Biol. 2016, 16, 259. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Roldán, J.L.; Dapporto, L.; Dincă, V.; Vicente, J.C.; Hornett, E.A.; Šíchová, J.; Lukhtanov, V.A.; Talavera, G.; Vila, R. Integrative analyses unveil speciation linked to host plant shift in Spialia butterflies. Mol. Ecol. 2016, 25, 4267–4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachos, F.E. An Annotated List of Species Concepts. In Species Concepts in Biology; Springer International Publishing: Cham, Switzerland, 2016; pp. 77–96. [Google Scholar]
- Mallet, J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 2005, 20, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Descimon, H.; Mallett, J. Bad Species. In Ecology of Butterflies in Europe; Settele, J., Shreeve, T., Konvič, M., Van Dyck, H., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 219–249. [Google Scholar]
- Mallet, J.; Beltrán, M.; Neukirchen, W.; Linares, M. Natural hybridization in heliconiine butterflies: The species boundary as a continuum. BMC Evol. Biol. 2007, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hausdorf, B. Progress toward a general species concept. Evol. Int. J. Org. Evol. 2011, 65, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Rundle, H.D.; Nosil, P. Ecological speciation. Ecol. Lett. 2005, 8, 336–352. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahami, M.S.; Dincă, V.; Lee, K.M.; Vila, R.; Joshi, M.; Heikkilä, M.; Dapporto, L.; Schmid, S.; Huemer, P.; Mutanen, M. Genomics Reveal Admixture and Unexpected Patterns of Diversity in a Parapatric Pair of Butterflies. Genes 2021, 12, 2009. https://doi.org/10.3390/genes12122009
Tahami MS, Dincă V, Lee KM, Vila R, Joshi M, Heikkilä M, Dapporto L, Schmid S, Huemer P, Mutanen M. Genomics Reveal Admixture and Unexpected Patterns of Diversity in a Parapatric Pair of Butterflies. Genes. 2021; 12(12):2009. https://doi.org/10.3390/genes12122009
Chicago/Turabian StyleTahami, Mohadeseh Sadat, Vlad Dincă, Kyung Min Lee, Roger Vila, Mukta Joshi, Maria Heikkilä, Leonardo Dapporto, Sarah Schmid, Peter Huemer, and Marko Mutanen. 2021. "Genomics Reveal Admixture and Unexpected Patterns of Diversity in a Parapatric Pair of Butterflies" Genes 12, no. 12: 2009. https://doi.org/10.3390/genes12122009
APA StyleTahami, M. S., Dincă, V., Lee, K. M., Vila, R., Joshi, M., Heikkilä, M., Dapporto, L., Schmid, S., Huemer, P., & Mutanen, M. (2021). Genomics Reveal Admixture and Unexpected Patterns of Diversity in a Parapatric Pair of Butterflies. Genes, 12(12), 2009. https://doi.org/10.3390/genes12122009