
Chromosome-Level Genome Assembly Provides New Insights into Genome Evolution and Tuberous Root Formation of Potentilla anserina

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and High-Throughput Sequencing
2.2. Estimation of Genome Size and Genome Assembly
2.3. Non-Coding RNA and Repeat Identification
2.4. Gene Prediction and Functional Annotation
2.5. Comparative Genomics and Phylogenetic Analysis
2.6. Whole Genome Duplication (WGD) Analysis
2.7. Sub-Genome Analysis and Expression Bias of Homeologs
2.8. Identification of Resistance Genes and Starch Biosynthesis Related Genes
3. Results
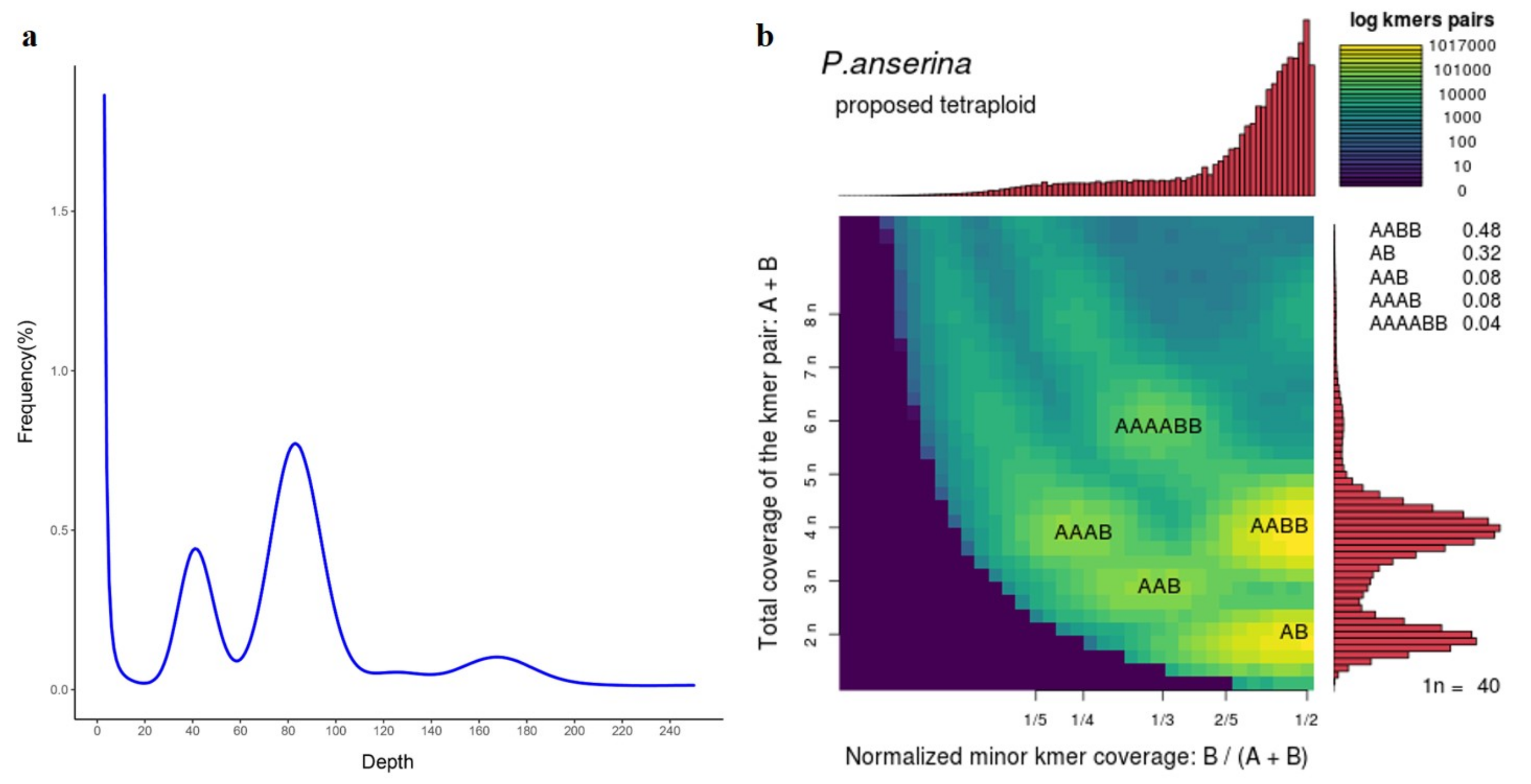
3.1. Genome Sequencing and Assembly
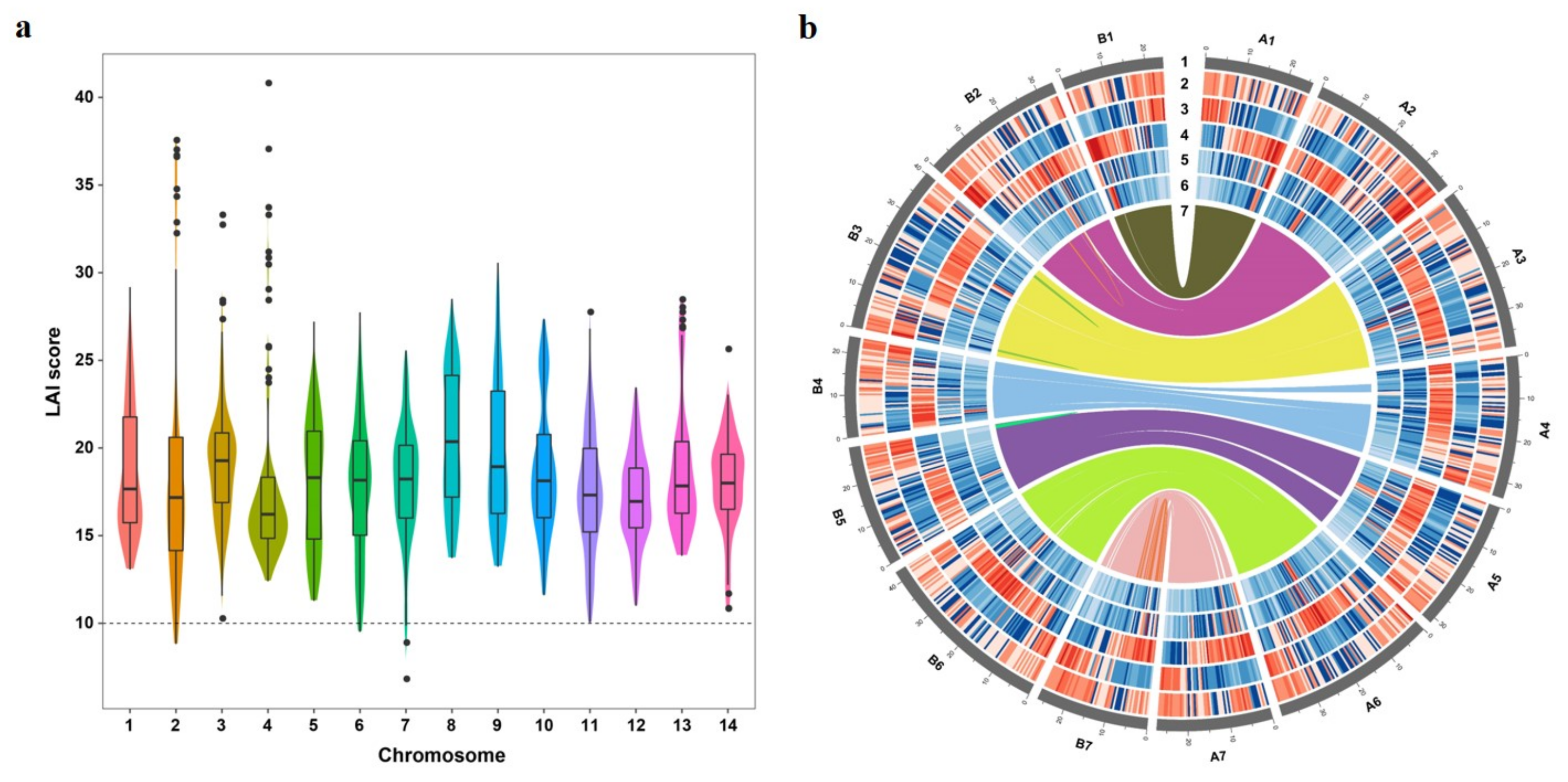
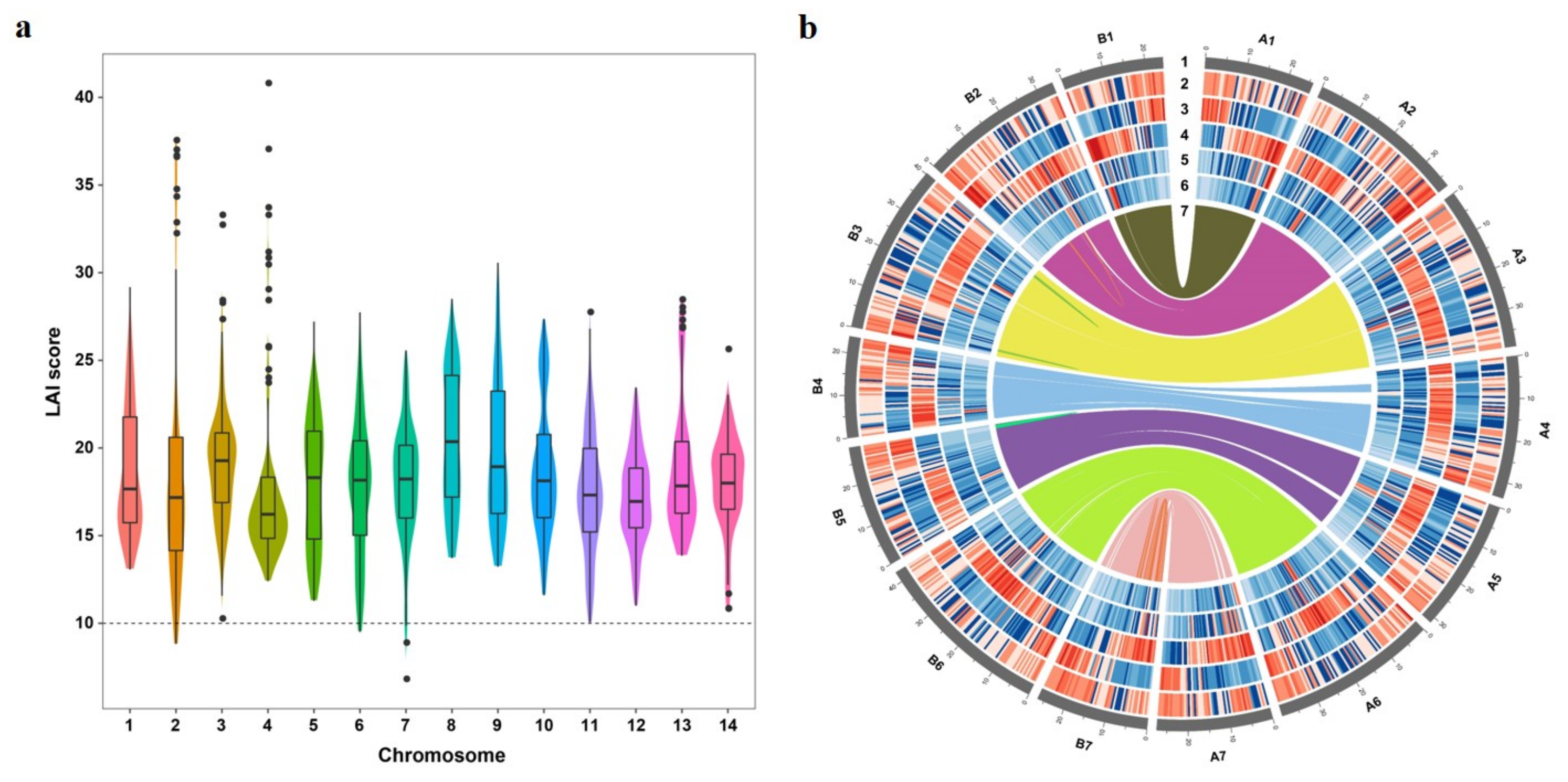
3.2. Assessment of Genome Quality
3.3. Genome Annotation
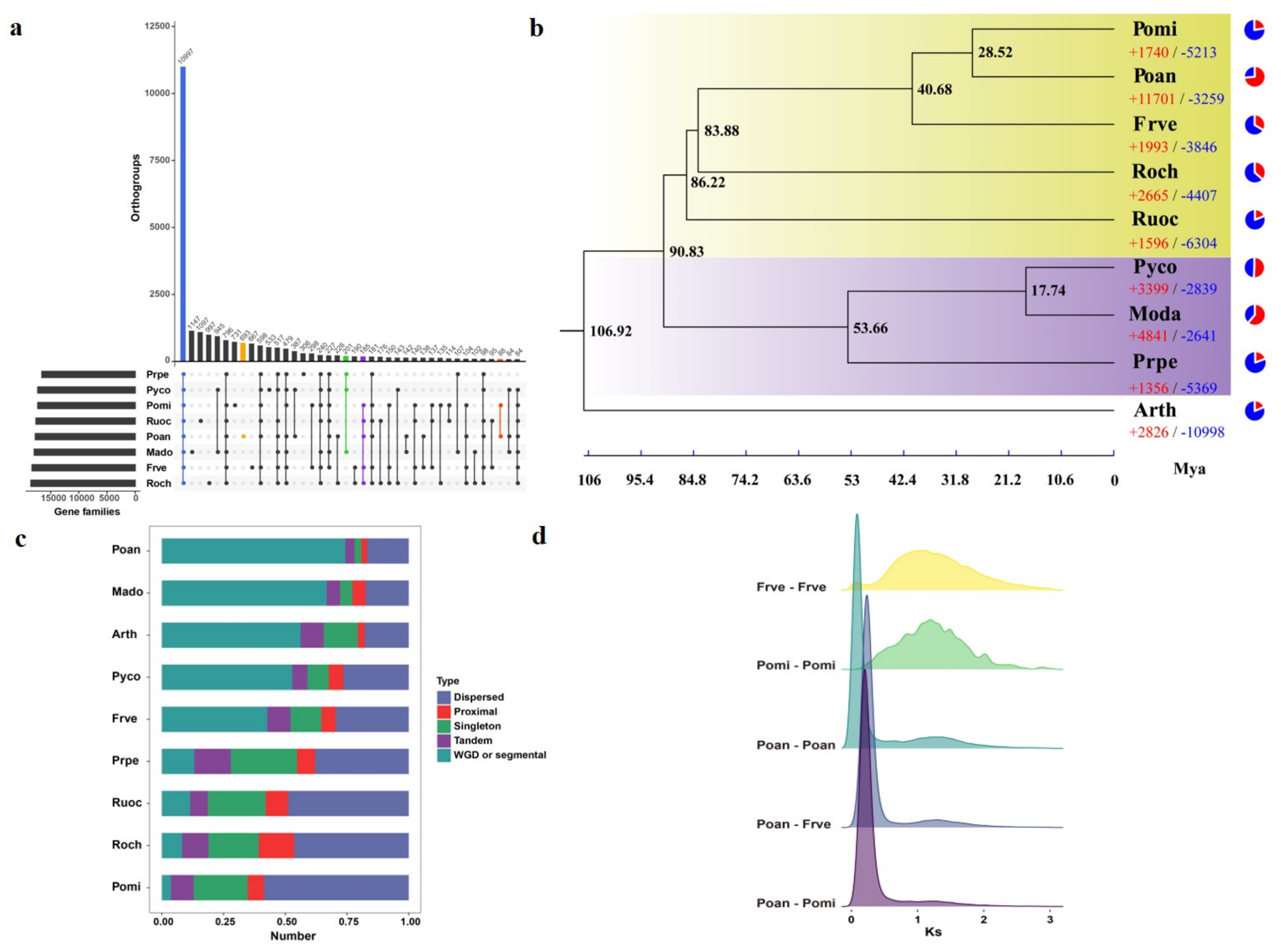
3.4. Comparative Genomics and Evolutionary Analysis
3.5. Sub-Genome Structure and Expression Bias Analysis
3.6. Resistance Gene Number Variation
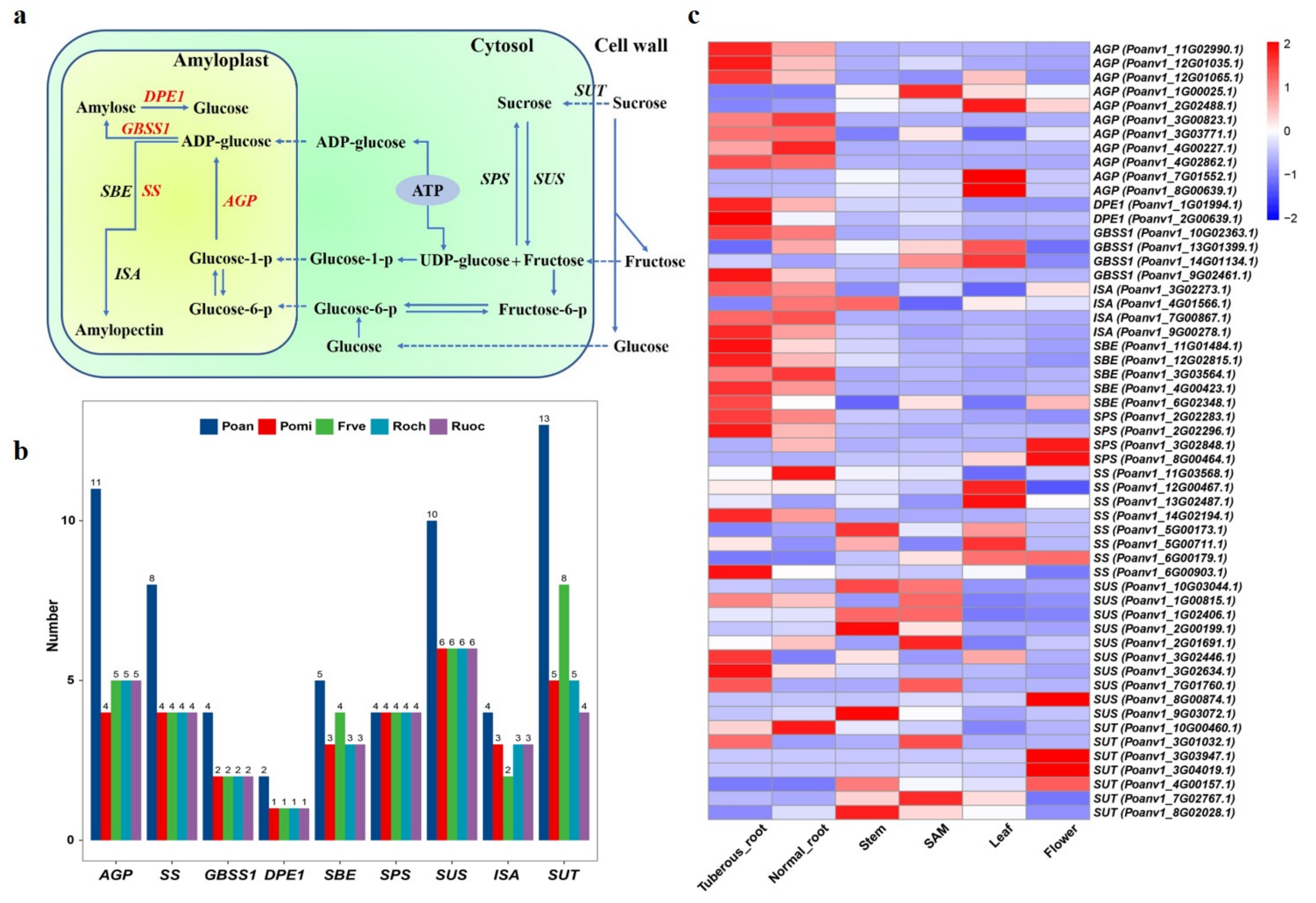
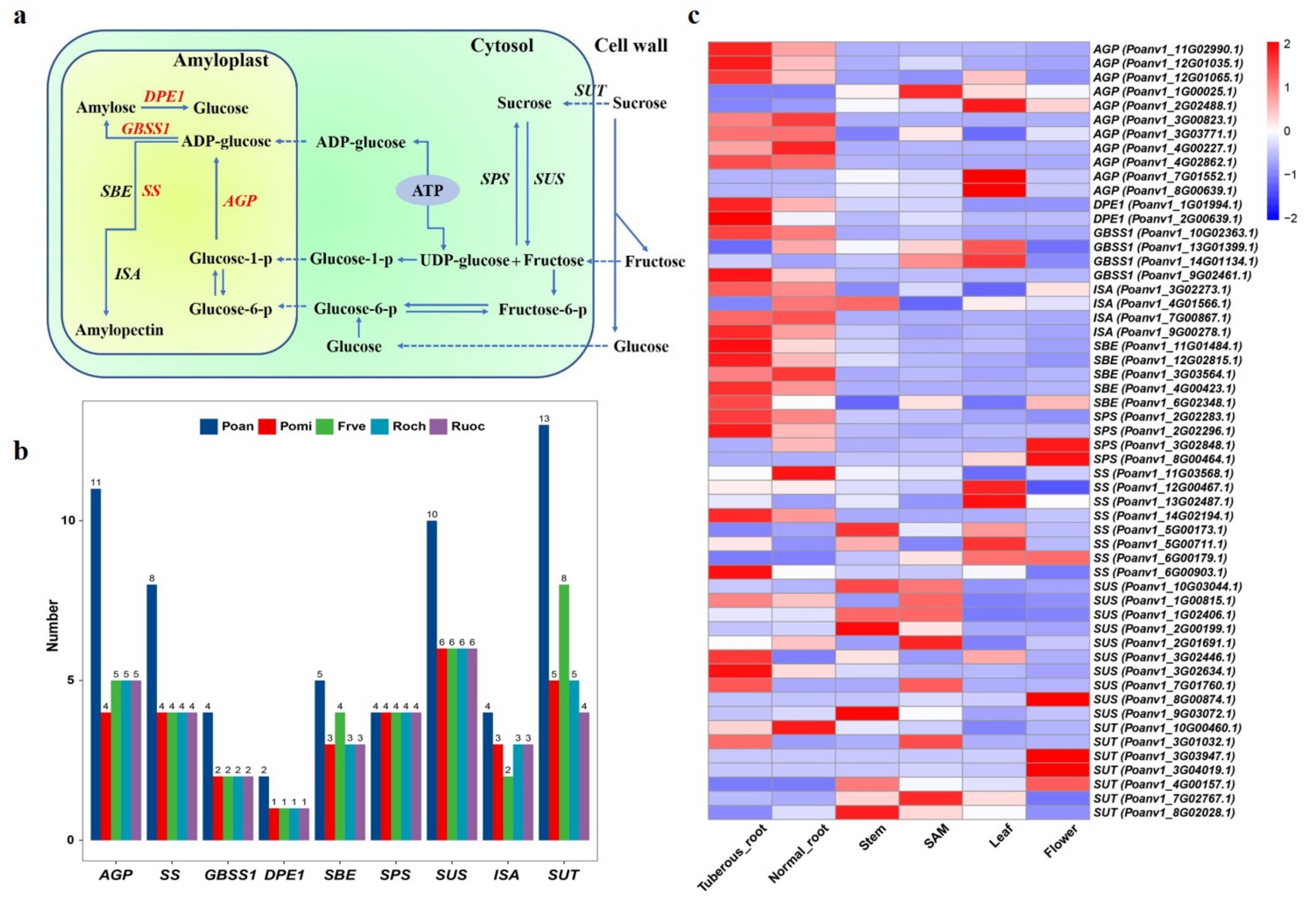
3.7. Genes Involved in Starch Biosynthesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Morikawa, T.; Imura, K.; Akagi, Y.; Muraoka, O.; Ninomiya, K. Ellagic acid glycosides with hepatoprotective activity from traditional Tibetan medicine Potentilla anserina. J. Nat. Med. 2017, 72, 29018991. [Google Scholar] [CrossRef] [PubMed]
- Northwest Plateau Institute of Biology, Chinese Academy of Sciences. Economic Flora of Qinghai; Qinghai Peoples Publishing House: Xining, China, 1987; pp. 270–273. [Google Scholar]
- Xia, L.; You, J. The determination of amino acids composition of the traditional food Potentilla anserina L. root by high-performance liquid chromatography via fluorescent detection and mass spectrometry. Int. J. Food Sci. Technol. 2011, 46, 1164–1170. [Google Scholar] [CrossRef]
- Guo, T.; Wei, J.; Ma, J. Antitussive and expectorant activities of Potentilla anserina. Pharm. Biol. 2015, 54, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Schimmer, O.; Lindenbaum, M. Tannins with Antimutagenic Properties in the Herb of Alchemilla Species and Potentilla anserina. Planta Med. 1995, 61, 141–145. [Google Scholar] [CrossRef]
- Kombal, R.; Glasl, H. Flavan-3-Ols and flavonoids from Potentilla anserina. Planta Med. 1995, 61, 484–485. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Cai, G.M.; Hong, X.; Shan, L.M.; Xiao, X. Anti-hepatitis B virus activities of triterpenoid saponin compound from Potentilla anserina L. Phytomedicine 2008, 15, 253–258. [Google Scholar] [CrossRef]
- Chen, J.R.; Yang, Z.Q.; Hu, T.J.; Yan, Z.T.; Niu, T.X.; Wang, L.; Cui, D.A.; Wang, M. Immunomodulatory activity in vitro and in vivo of polysaccharide from Potentilla anserina. Fitoterapia 2010, 81, 1117–1124. [Google Scholar] [CrossRef]
- Eriksson, T.; Donoghue, M.; Hibbs, M. Phylogenetic analysis of Potentilla using DNA sequences of nuclear ribosomal internal transcribed spacers (ITS), and implications for the classification of Rosoideae (Rosaceae). Plant Syst. Evol. 1998, 211, 155–179. [Google Scholar] [CrossRef]
- Persson, N.; Toresen, I.; Andersen, H.; Smedmark, J.; Eriksson, T. Detecting destabilizing species in the phylogenetic backbone of Potentilla (Rosaceae) using low-copy nuclear markers. AoB Plants 2020, 12, plaa017. [Google Scholar] [CrossRef] [PubMed]
- Töpel, M.; Lundberg, M.; Eriksson, T.; Eriksen, B. Molecular data and ploidal levels indicate several putative allopolyploidization events in the genus Potentilla (Rosaceae). PLoS Curr. 2011, 3, RRN1237. [Google Scholar] [CrossRef]
- Buti, M.; Moretto, M.; Barghini, E.; Mascagni, F.; Natali, L.; Brilli, M.; Lomsadze, A.; Sonego, P.; Giongo, L.; Alonge, M.; et al. The genome sequence and transcriptome of Potentilla micrantha and their comparison to Fragaria vesca (the woodland strawberry). GigaScience 2018, 7, giy010. [Google Scholar] [CrossRef] [Green Version]
- Ranallo-Benavidez, T.; Jaron, K.; Schatz, M. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.; Nattestad, M.; Concepcion, G.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.; Zeng, Q.; Wortman, J.; Young, S.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef] [Green Version]
- Dudchenko, O.; Batra, S.; Omer, A.; Nyquist, S.; Hoeger, M.; Durand, N.; Shamim, S.; Machol, I.; Lander, E.; Aiden, A.; et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, eaal3327. [Google Scholar] [CrossRef] [Green Version]
- Durand, N.; Shamim, S.; Machol, I.; Rao, S.; Huntley, M.; Lander, E.; Aiden, E. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.; Waterhouse, R.; Ioannidis, P.; Zdobnov, E. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Chen, J.; Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 2018, 46, e126. [Google Scholar] [CrossRef]
- Ou, S.; Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.; Eddy, S. tRNAscan-SE: A program for improved detection of transfer RNA Genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, E.; Kolbe, D.; Eddy, S. Infernal 1.0: Inference of RNA Alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalvari, I.; Nawrocki, E.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2020, 49, D192–D200. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.; Feschotte, C.; Smit, A. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 201921046. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR-FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [Green Version]
- Jurka, J.; Kapitonov, V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 9, 1–9. [Google Scholar]
- Goodstein, D.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2011, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Birney, E.; Durbin, R. Using GeneWise in the Drosophila Annotation Experiment. Genome Res. 2000, 10, 547–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.; Salzberg, S.; Zhu, W.; Pertea, M.; Allen, J.; Orvis, J.; White, O.; Buell, C.; Wortman, J. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.; Luciani, A.; Potter, S.; Qureshi, M.; Richardson, L.; Salazar, G.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Lee, T.; Cheng, C.H.; Buble, K.; Zheng, P.; Yu, J.; Humann, J.; Ficklin, S.; Gasic, K.; Scott, K.; et al. 15 years of GDR: New data and functionality in the Genome Database for Rosaceae. Nucleic Acids Res. 2018, 47, D1137–D1145. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2016, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Dechao, B.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Lianhe, Z.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Edgar, R. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.; Clements, J.; Eddy, S. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.; Park, C.; Bennett, C.; Salzberg, S. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 1. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, D.; Ho, Y.S.; Lightfoot, D.; Schmöckel, S.; Li, B.; Borm, T.; Ohyanagi, H.; Mineta, K.; Michell, C.; Saber, N.; et al. The genome of Chenopodium quinoa. Nature 2017, 542, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Yasui, Y.; Hirakawa, H.; Oikawa, T.; Toyoshima, M.; Matsuzaki, C.; Ueno, M.; Mizuno, N.; Nagatoshi, Y.; Imamura, T.; Miyago, M.; et al. Draft genome sequence of an inbred line of Chenopodium quinoa, an allotetraploid crop with great environmental adaptability and outstanding nutritional properties. DNA Res. 2016, 23, dsw037. [Google Scholar] [CrossRef] [Green Version]
- Sahebi, M.; Hanafi, M.; Wijnen, A.; Rice, D.; Rafii, M.; Azizi, P.; Osman, M.; Taheri, S.; Abu Bakar, M.F.; Mat Isa, M.N.; et al. Contribution of transposable elements in the plant’s genome. Gene 2018, 665, 155–166. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Li, W.; Li, K.; Nan, H.; Shi, C.; Zhang, Y.; Dai, Z.Y.; Lin, Y.L.; Yang, X.L.; Tong, Y.; et al. The Chromosome-Level Reference Genome of Tea Tree Unveils Recent Bursts of Non-autonomous LTR Retrotransposons to Drive Genome Size Evolution. Mol. Plant 2020, 13, 935–938. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Gao, L.Z. Rapid and Recent Evolution of LTR Retrotransposons Drives Rice Genome Evolution During the Speciation of AA- Genome Oryza Species. G3 Bethesda Md. 2017, 7, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.H. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.T.; Wang, R.; Zhang, S.; Paterson, A. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, R.; Wu, X.; Wang, J. Homoeolog expression bias and expression level dominance (ELD) in four tissues of natural allotetraploid Brassica napus. BMC Genom. 2020, 21, 330. [Google Scholar] [CrossRef]
- Lozano, R.; Hamblin, M.; Prochnik, S.; Jannink, J.L. Identification and distribution of the NBS-LRR gene family in the Cassava genome. BMC Genom. 2015, 16, 360. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yao, W.; Wang, J.; Jidong, W.; Ma, H.; Zhang, Y. A novel effect of glycine on the growth and starch biosynthesis of storage root in sweetpotato (Ipomoea batatas Lam.). Plant Physiol. Biochem. 2019, 144, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Abt, M.; Zeeman, S. Evolutionary innovations in starch metabolism. Curr. Opin. Plant Biol. 2020, 55, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Seung, D. Amylose in starch: Towards an understanding of biosynthesis, structure and function. New Phytol. 2020, 228, 1490–1504. [Google Scholar] [CrossRef] [PubMed]
- Utsumi, Y.; Tanaka, M.; Utsumi, C.; Takahashi, S.; Matsui, A.; Fukushima, A.; Kobayashi, M.; Sasaki, R.; Oikawa, A.; Kusano, M.; et al. Integrative omics approaches revealed a crosstalk among phytohormones during tuberous root development in cassava. Plant Mol. Biol. 2020, 8, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zierer, W.; Rüscher, D.; Sonnewald, U.; Sonnewald, S. Tuber and Tuberous Root Development. Annu. Rev. Plant Biol. 2021, 72, 551–580. [Google Scholar] [CrossRef] [PubMed]
- Cobon, A.M.; Matfield, B. Morphological and cytological studies on a hexaploid clone of Potentilla anserina L. Watsonia 1976, 11, 125–129. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Number | Total Length (bp) | Proportion in the Genome (%) | |
|---|---|---|---|---|
| miRNA | 479 | 58,847 | 0.013 | |
| tRNA | 1116 | 83,079 | 0.018 | |
| Non-coding RNA | rRNA | 148 | 98,151 | 0.022 |
| snRNA | 655 | 77,523 | 0.017 | |
| Total | 2398 | 317,600 | 0.07 | |
| Retroelements | 166,482 | 118,458,221 | 26.08 | |
| LINEs | 39,955 | 12,791,517 | 2.82 | |
| SINEs | 2373 | 269,946 | 0.06 | |
| LTR elements | 124,154 | 105,396,758 | 23.20 | |
| TE | Gypsy | 56,850 | 53,227,095 | 11.72 |
| Copia | 31,449 | 38,942,113 | 8.57 | |
| DNA transposons | 167,514 | 4,317,7431 | 9.50 | |
| Others | 27,886 | 6,454,876 | 1.42 | |
| Total | 368,838 | 169,737,457 | 37.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gan, X.; Li, S.; Zong, Y.; Cao, D.; Li, Y.; Liu, R.; Cheng, S.; Liu, B.; Zhang, H. Chromosome-Level Genome Assembly Provides New Insights into Genome Evolution and Tuberous Root Formation of Potentilla anserina. Genes 2021, 12, 1993. https://doi.org/10.3390/genes12121993
Gan X, Li S, Zong Y, Cao D, Li Y, Liu R, Cheng S, Liu B, Zhang H. Chromosome-Level Genome Assembly Provides New Insights into Genome Evolution and Tuberous Root Formation of Potentilla anserina. Genes. 2021; 12(12):1993. https://doi.org/10.3390/genes12121993
Chicago/Turabian StyleGan, Xiaolong, Shiming Li, Yuan Zong, Dong Cao, Yun Li, Ruijuan Liu, Shu Cheng, Baolong Liu, and Huaigang Zhang. 2021. "Chromosome-Level Genome Assembly Provides New Insights into Genome Evolution and Tuberous Root Formation of Potentilla anserina" Genes 12, no. 12: 1993. https://doi.org/10.3390/genes12121993
APA StyleGan, X., Li, S., Zong, Y., Cao, D., Li, Y., Liu, R., Cheng, S., Liu, B., & Zhang, H. (2021). Chromosome-Level Genome Assembly Provides New Insights into Genome Evolution and Tuberous Root Formation of Potentilla anserina. Genes, 12(12), 1993. https://doi.org/10.3390/genes12121993