
The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
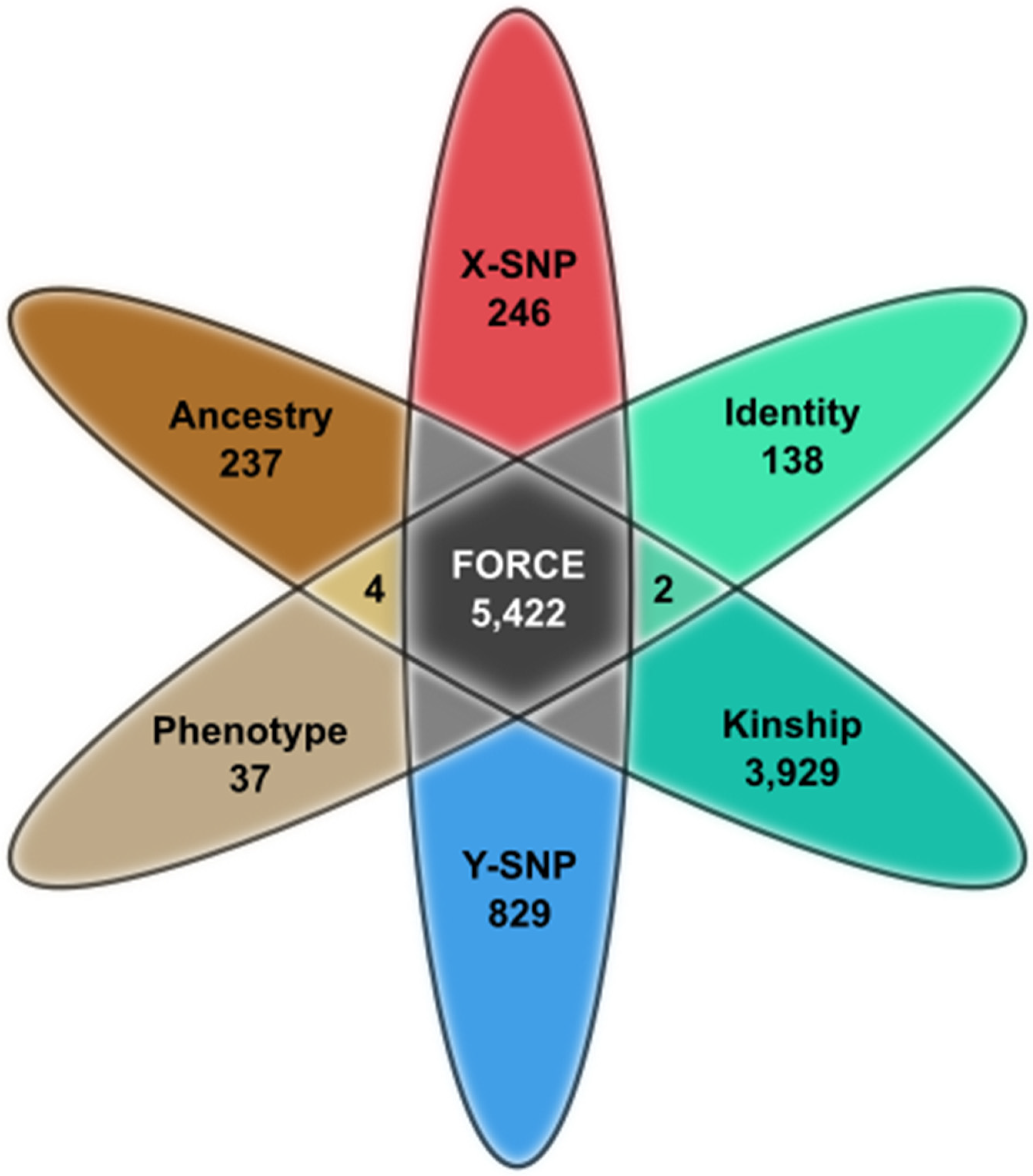
2.1. FORCE Panel Design
2.2. Simulated Panel Performance

2.3. SNP Capture Assay Laboratory Testing
2.3.1. Sample Selection
2.3.2. DNA Extraction and Quantitation
2.3.3. Library Preparation
2.3.4. Hybridization Capture
2.3.5. Normalization, Pooling, and High Throughput Sequencing
2.3.6. Sequence Data Analysis
2.3.7. SNP Concordance Assessment
2.3.8. Biogeographical Ancestry Predictions
2.3.9. Phenotype Predictions
2.3.10. Y Haplogroup Predictions
2.3.11. Kinship Statistics (Kinship SNP and X-SNP)
3. Results and Discussion
3.1. Simulated Panel Performance
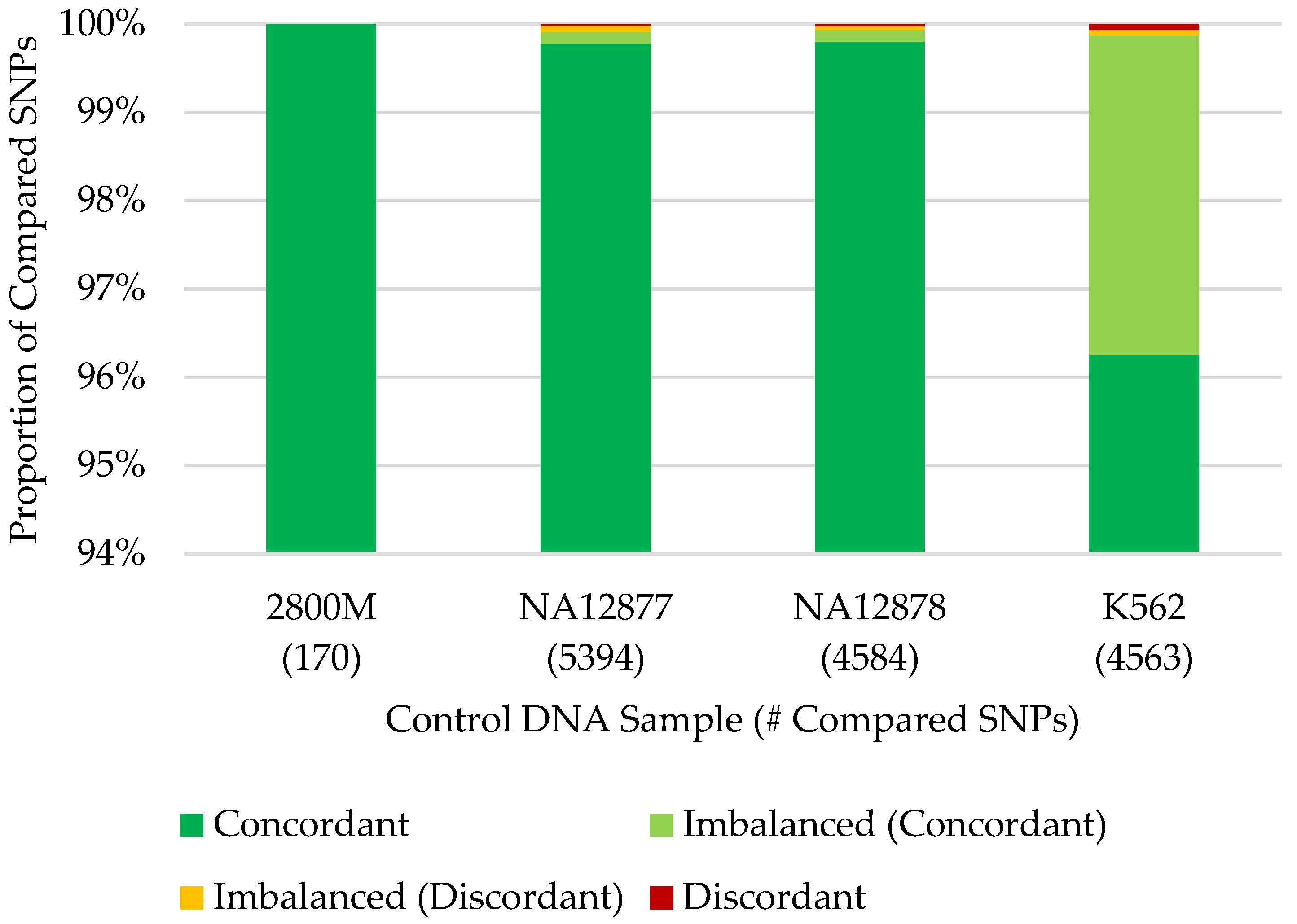
3.2. SNP Capture Assay Laboratory Testing
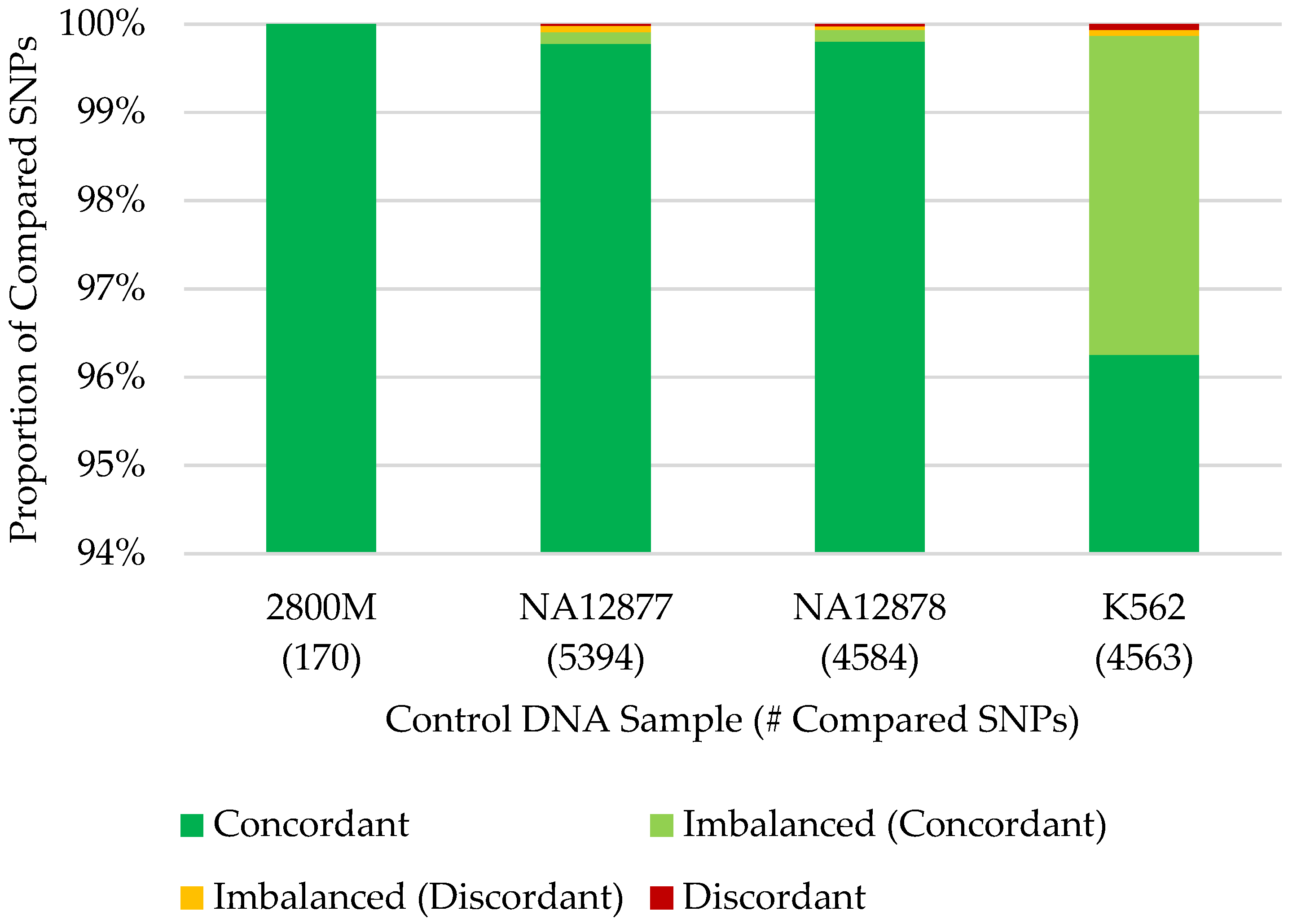
3.3. Concordance with Published SNP Data
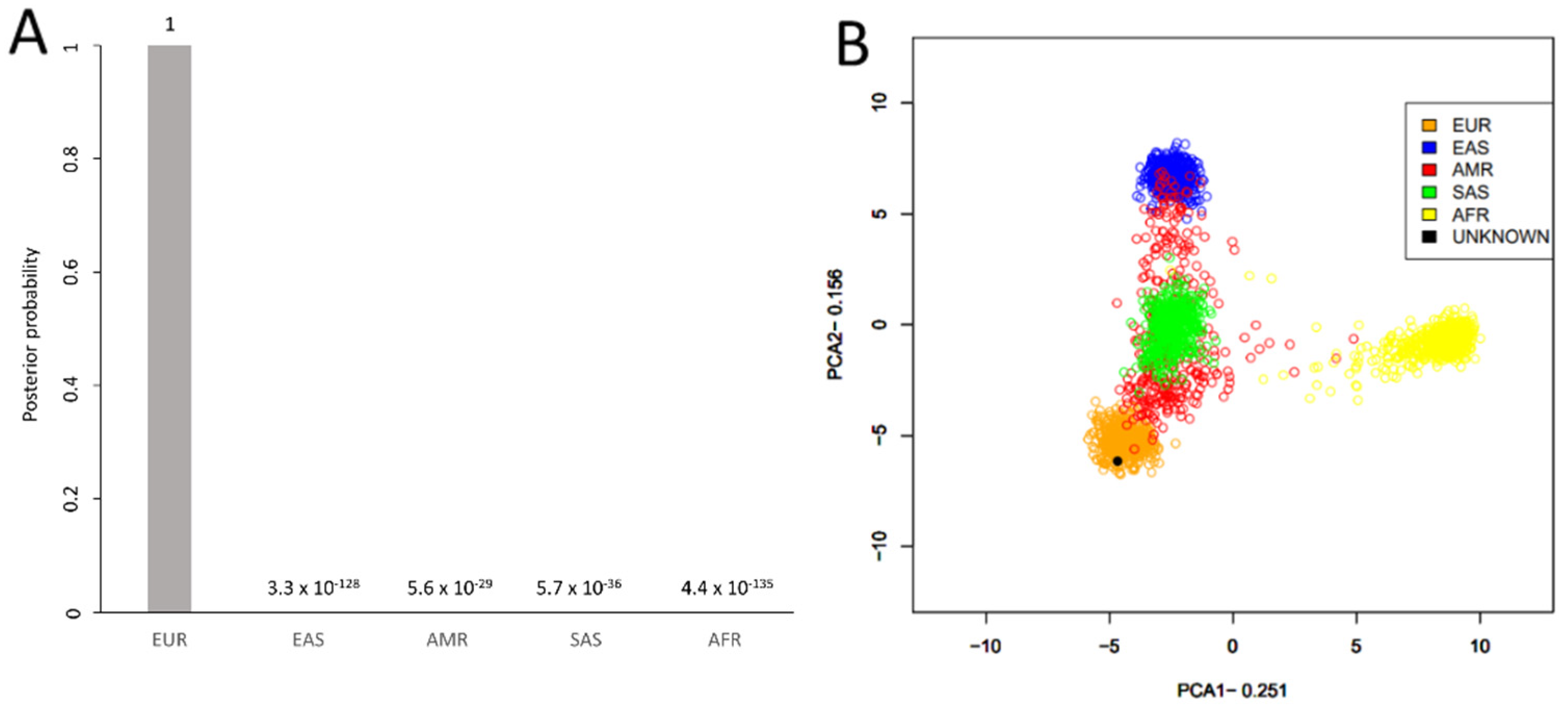
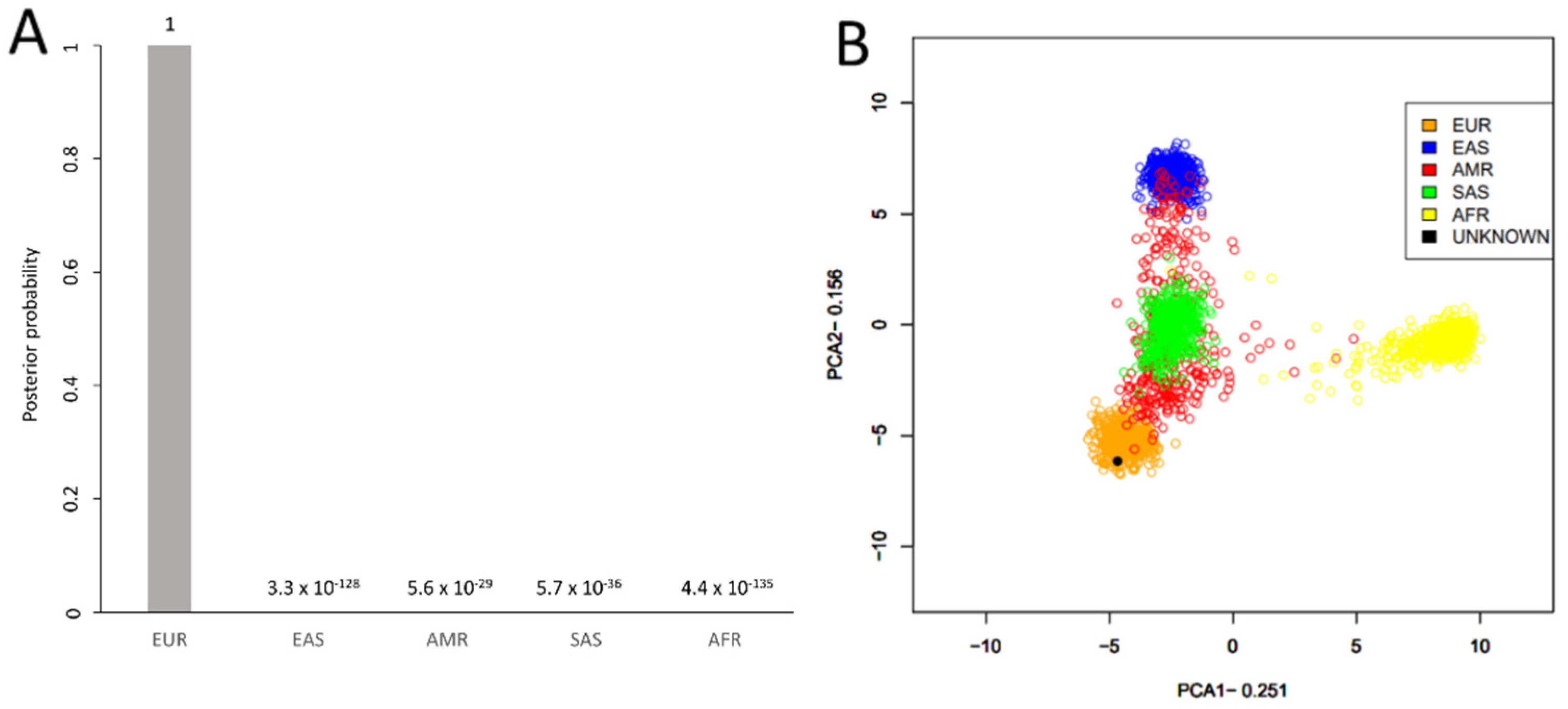
3.4. Ancestry, Phenotype, and Y Haplogroup Predictions
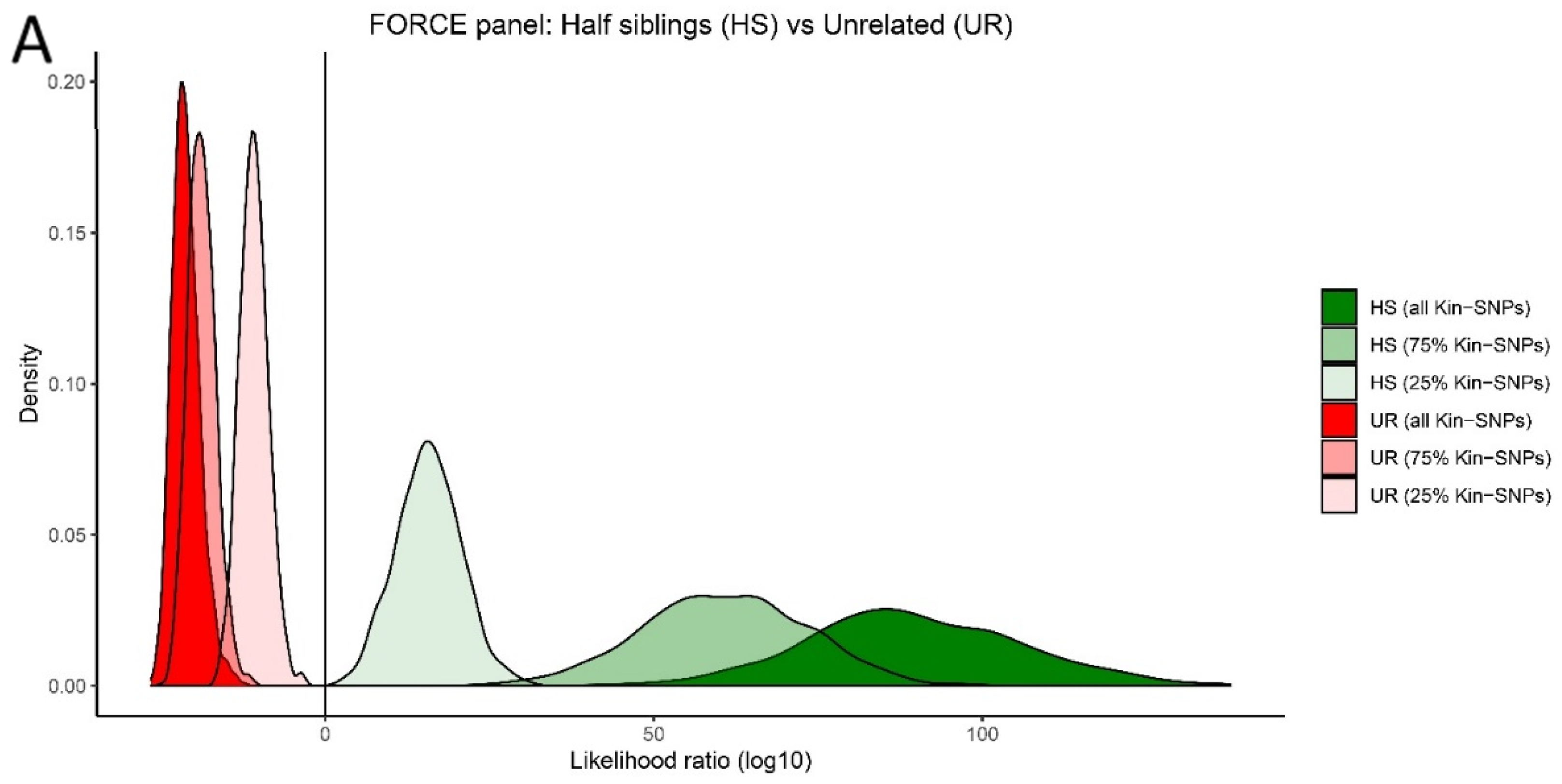
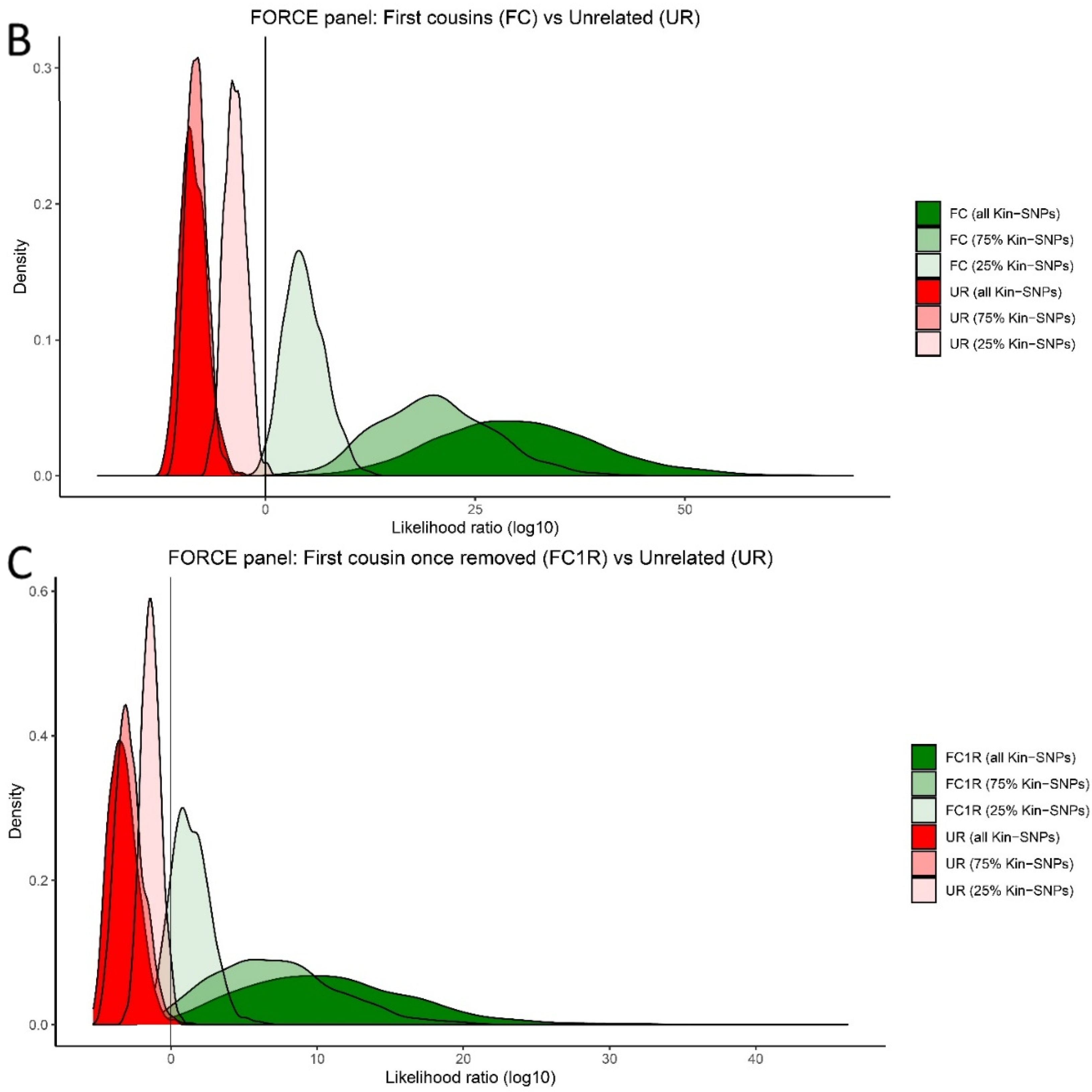
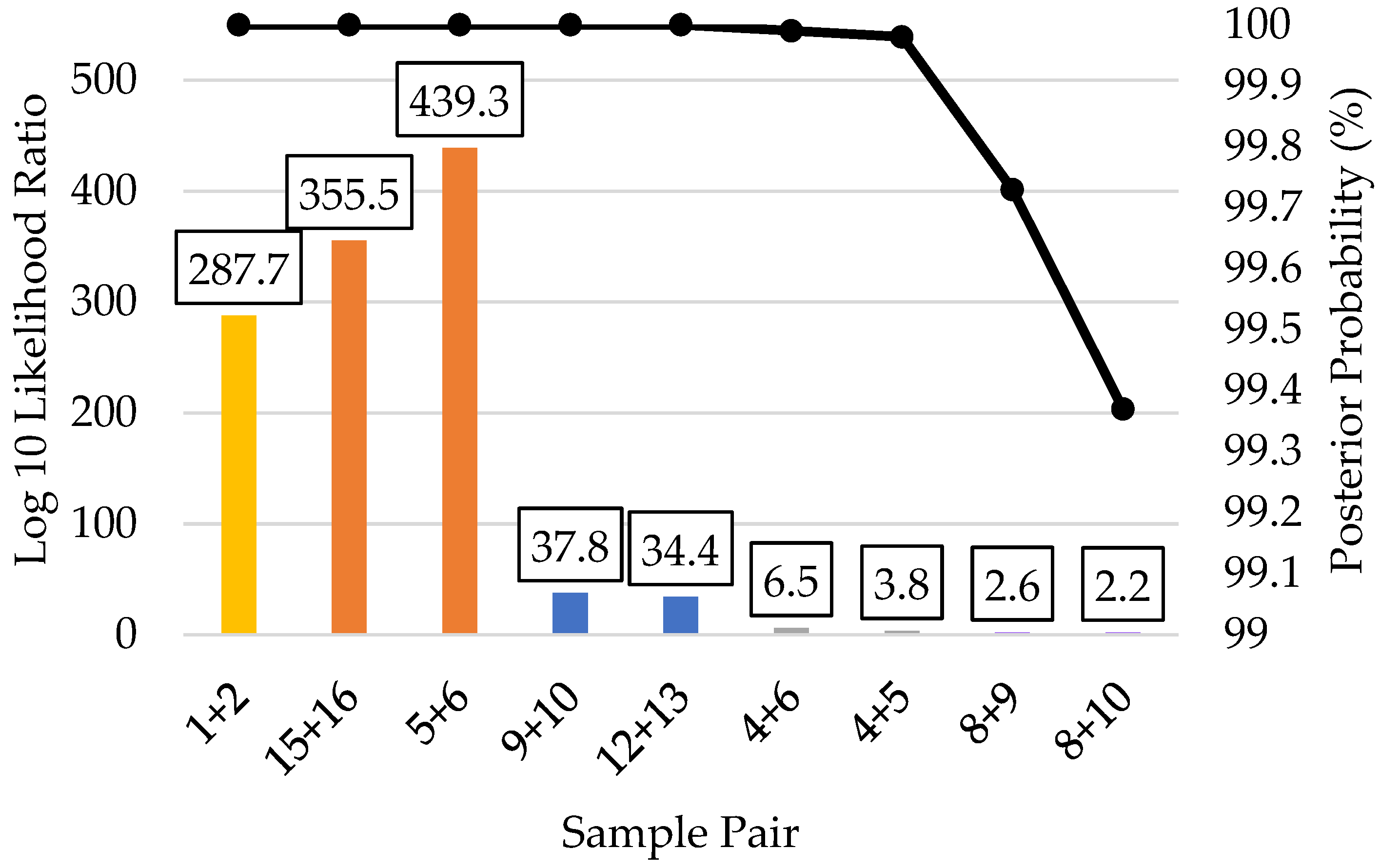
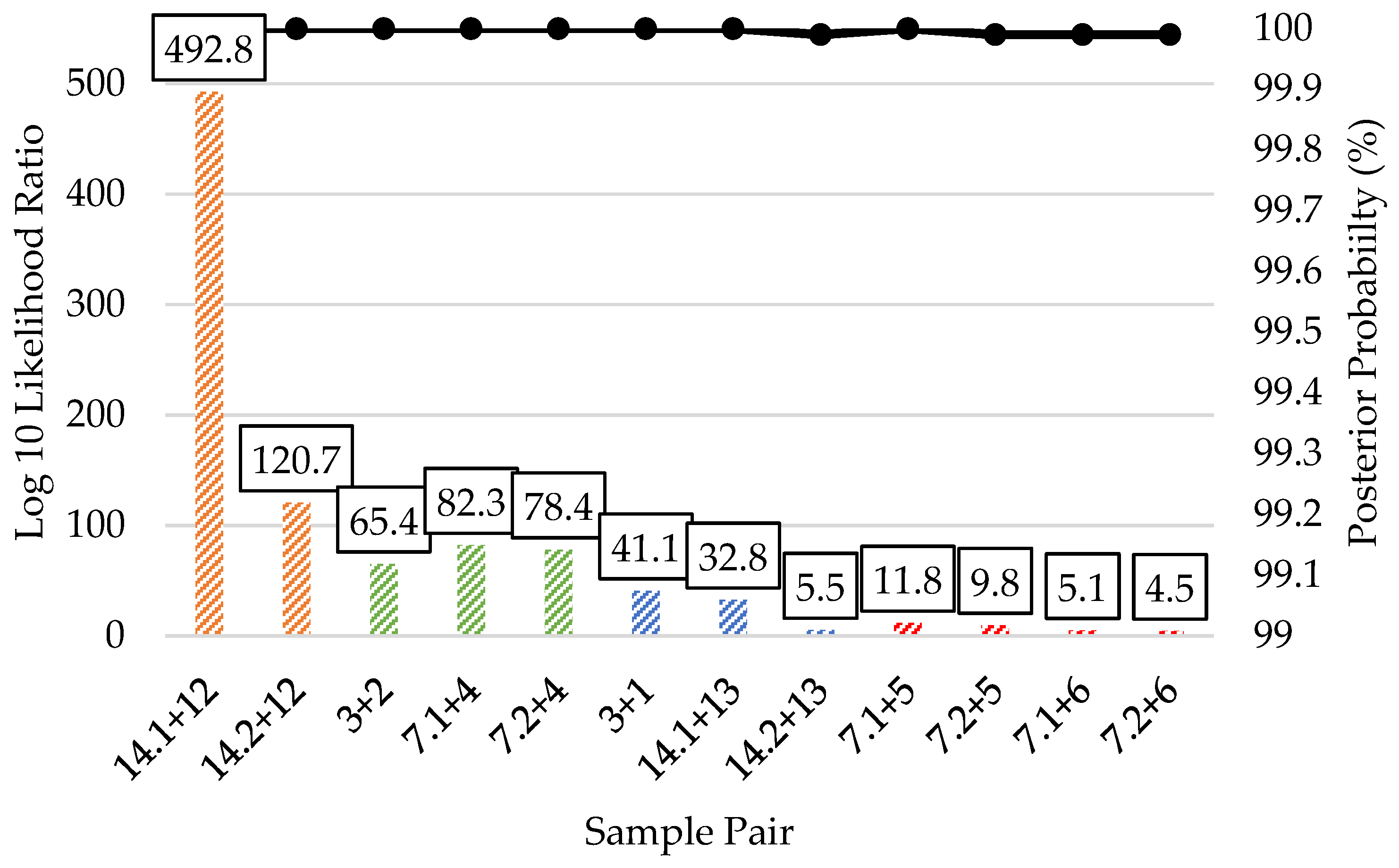
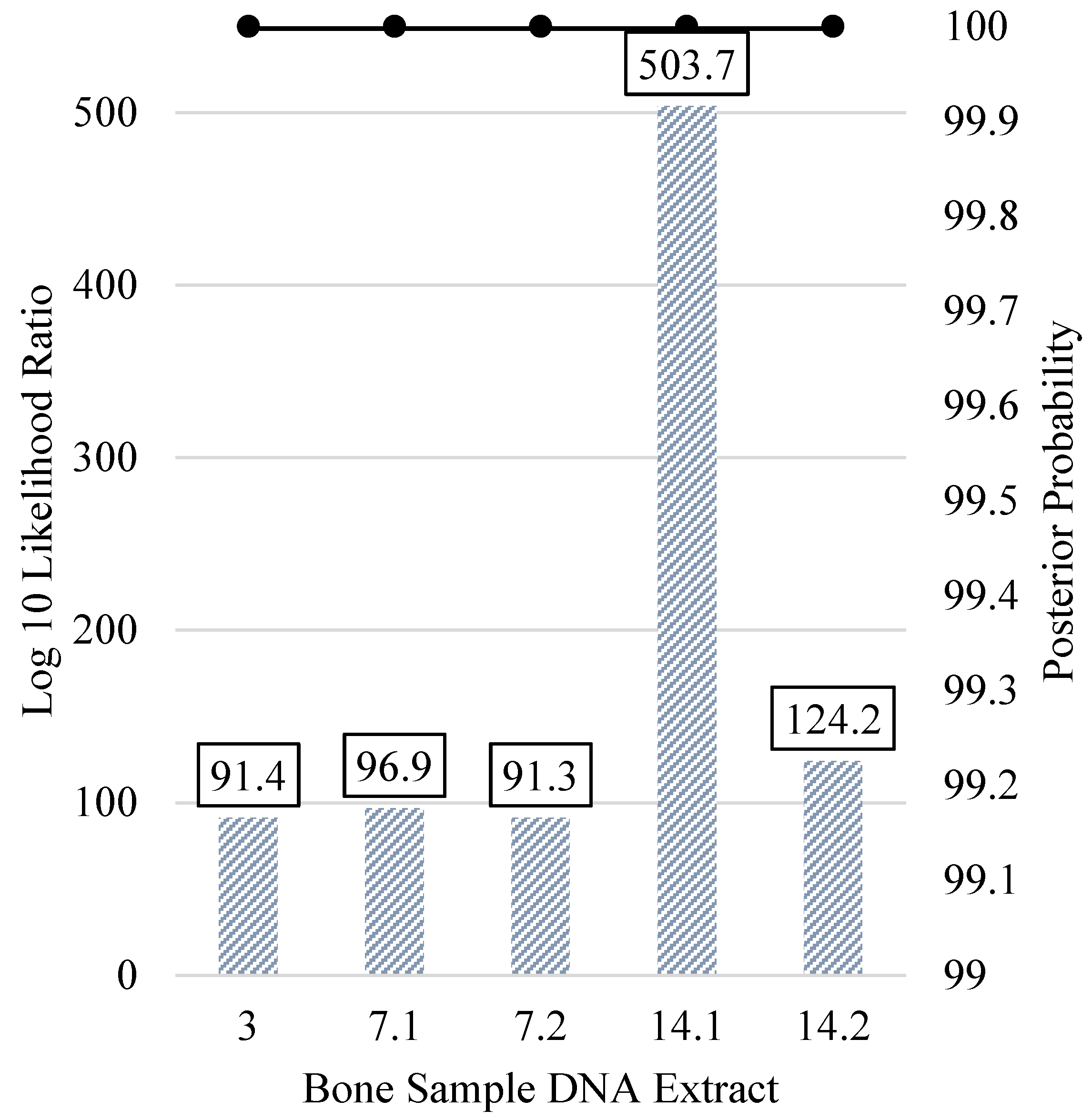
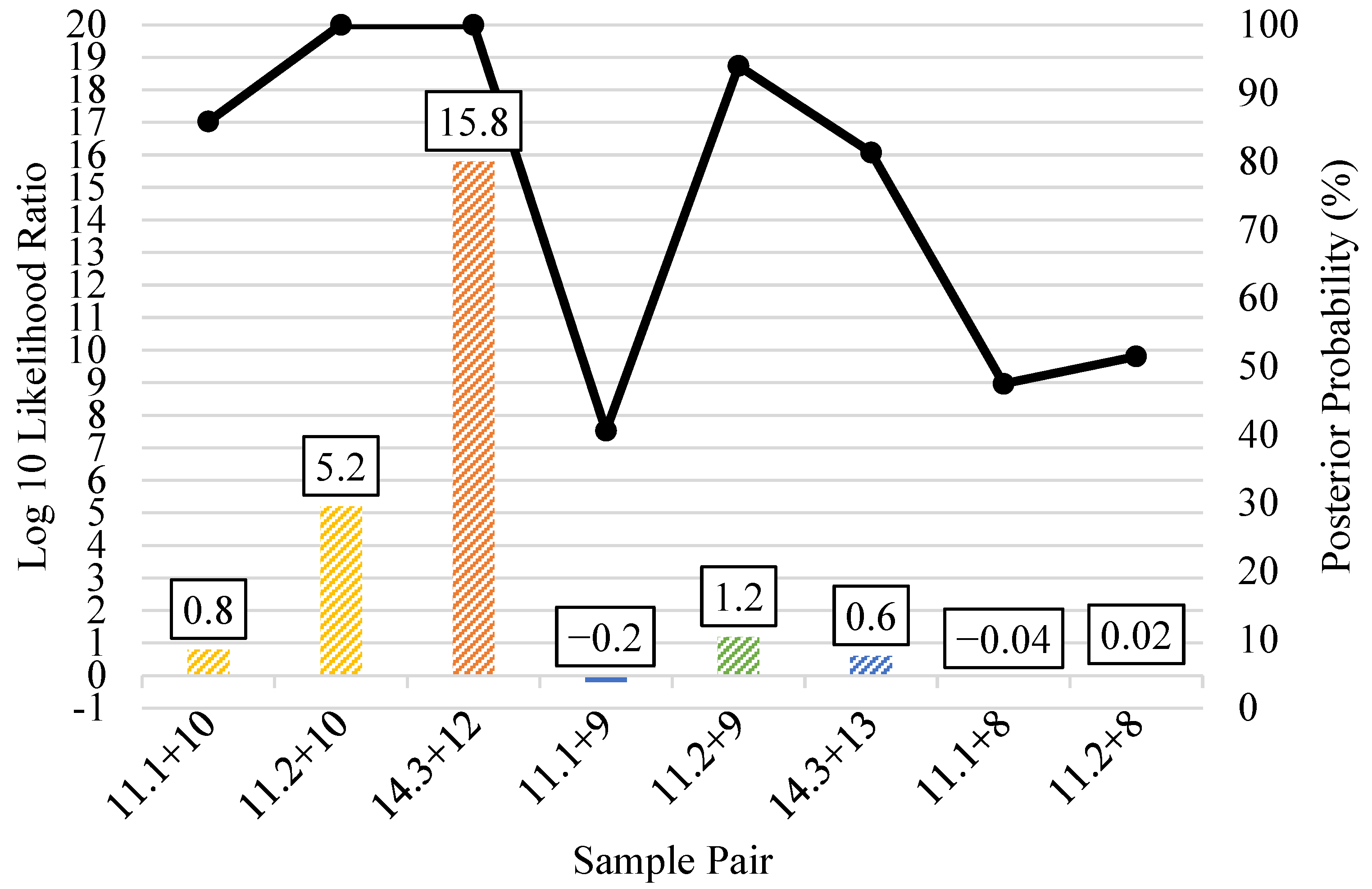
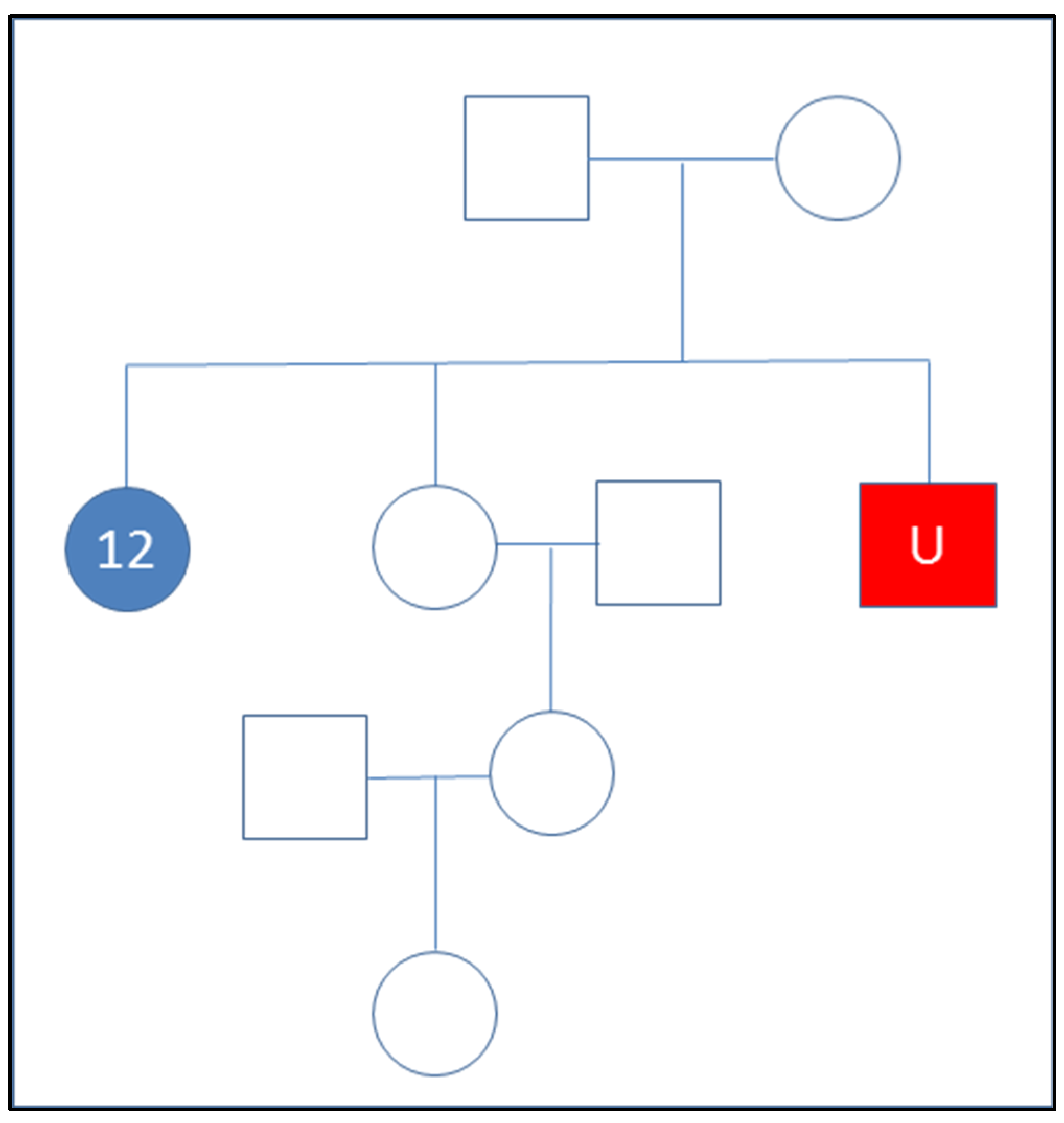
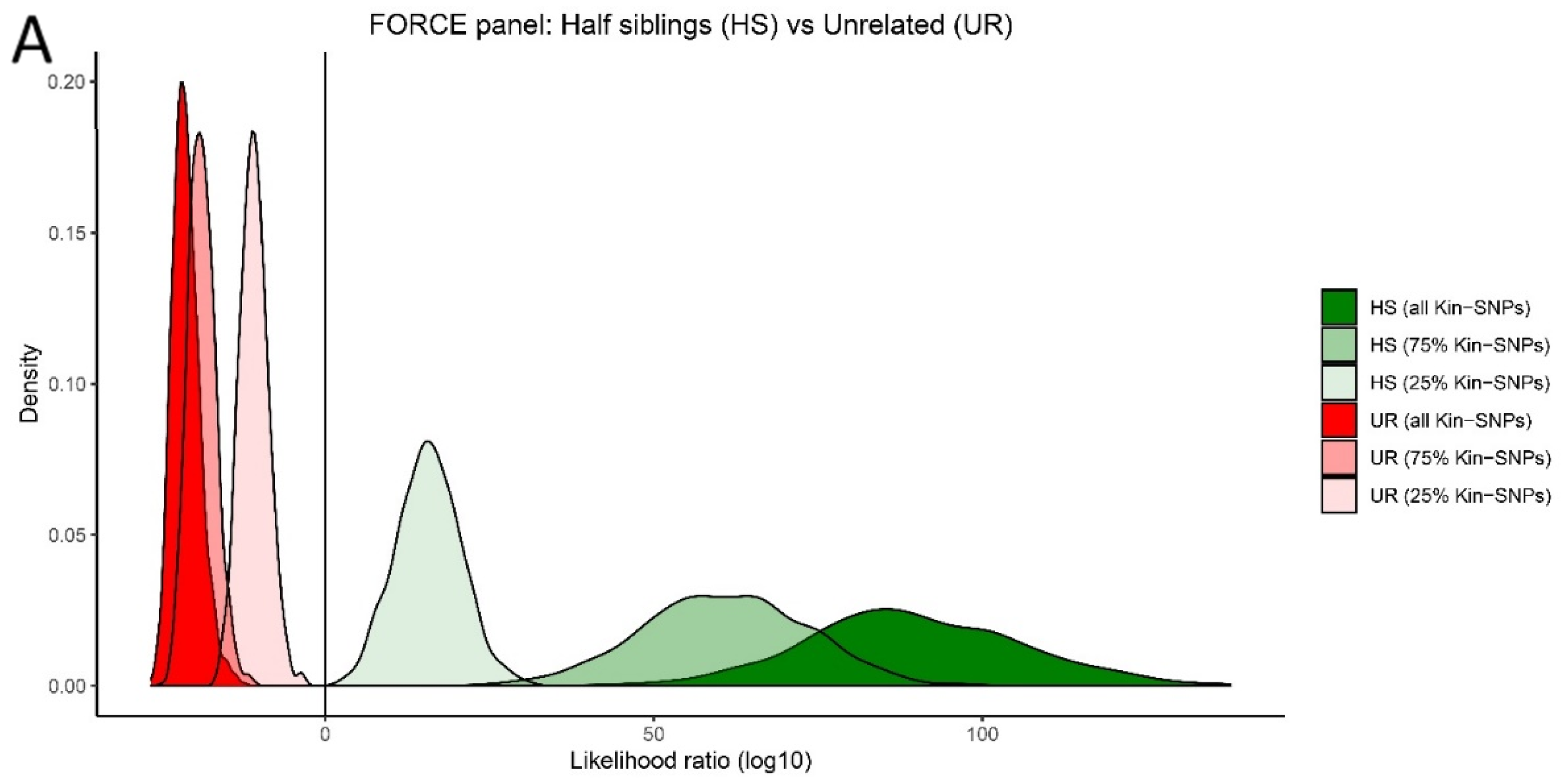
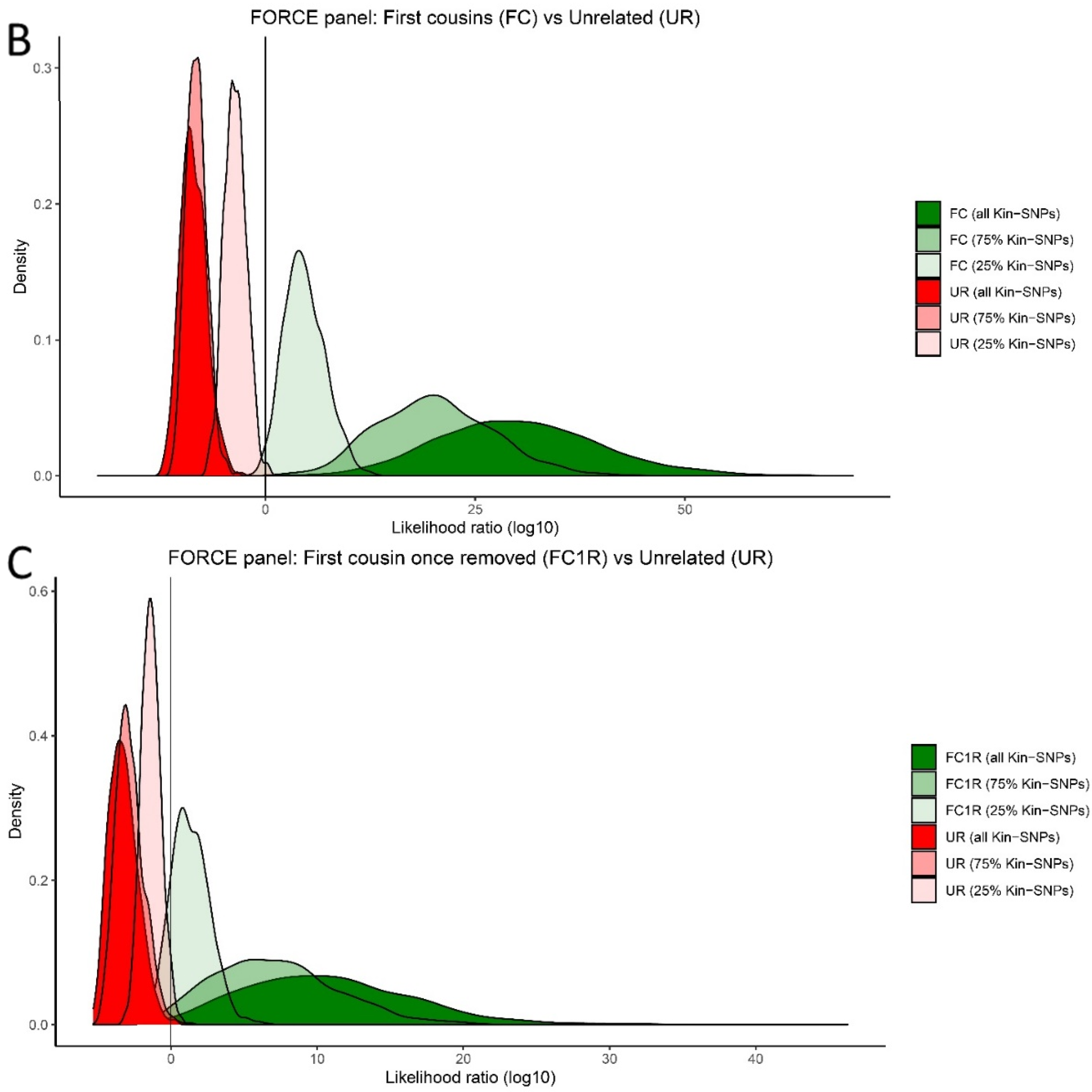
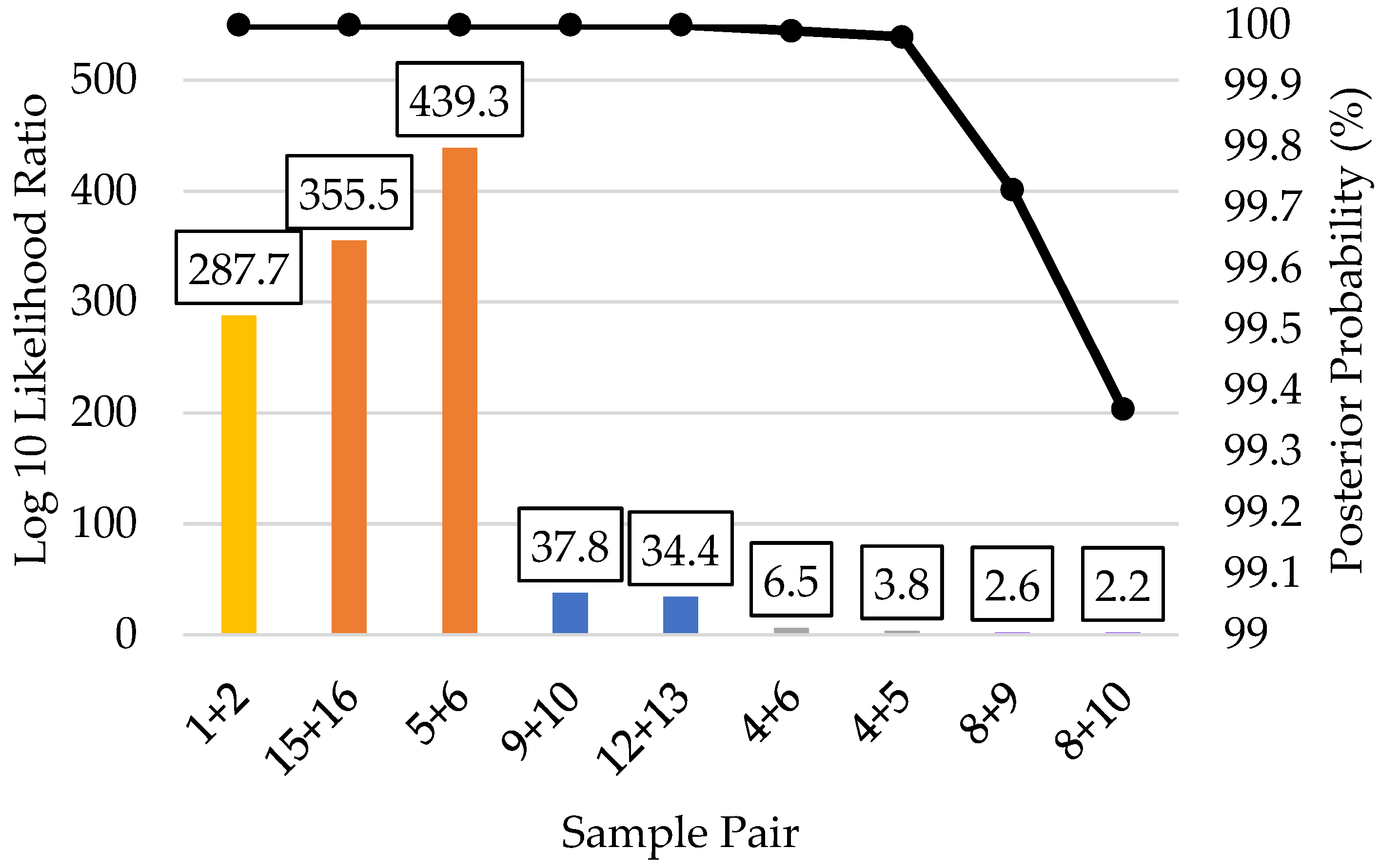
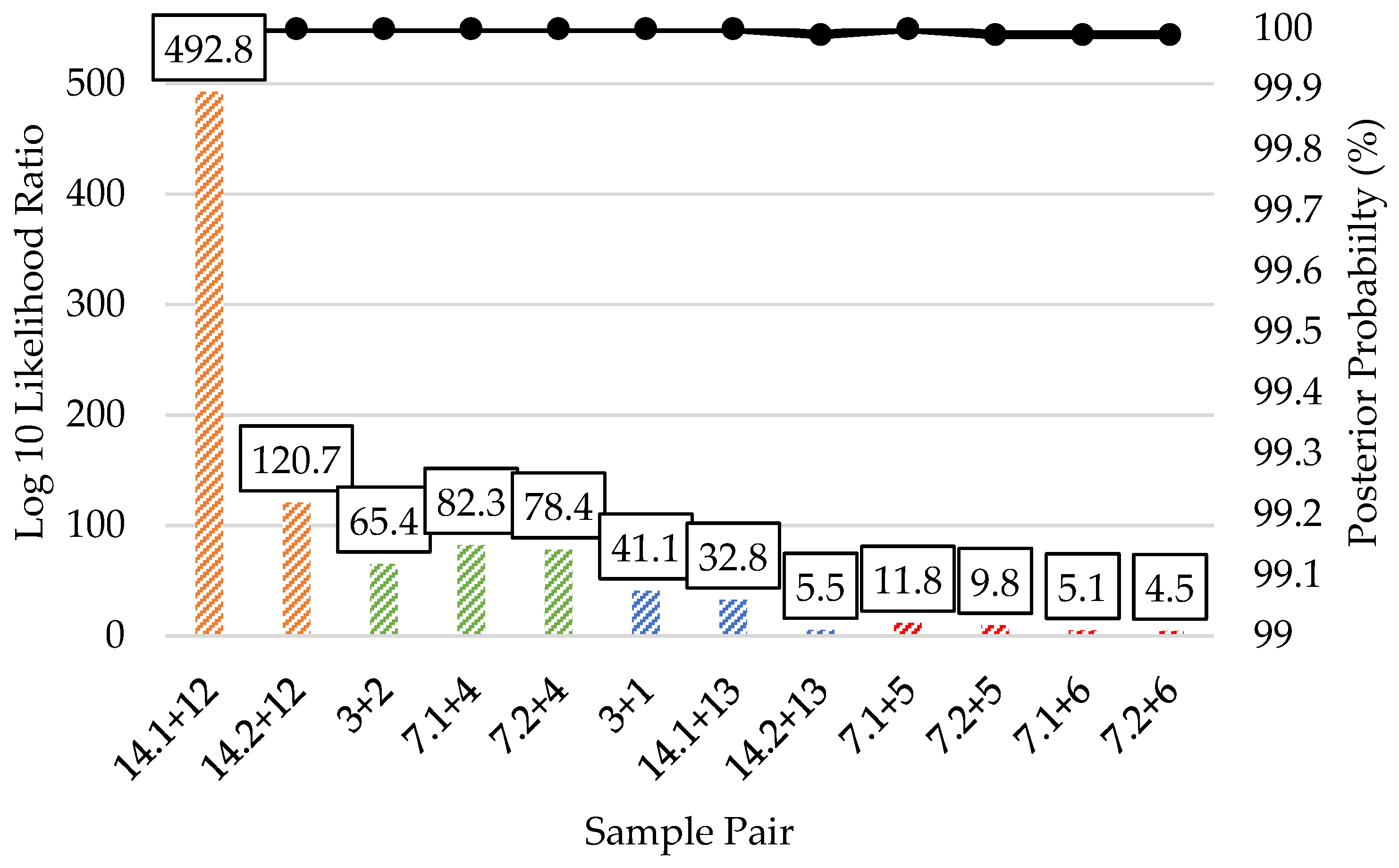
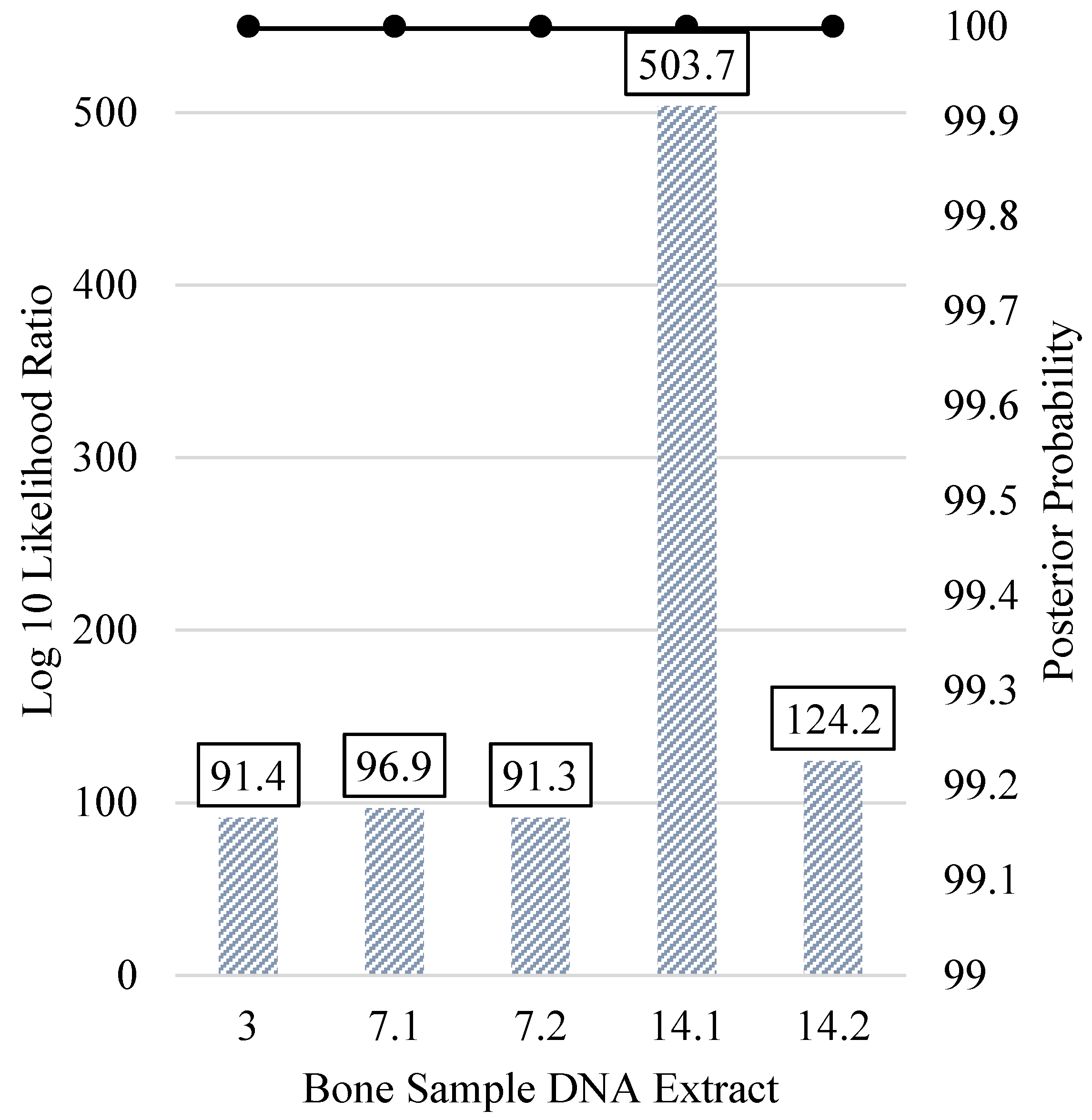
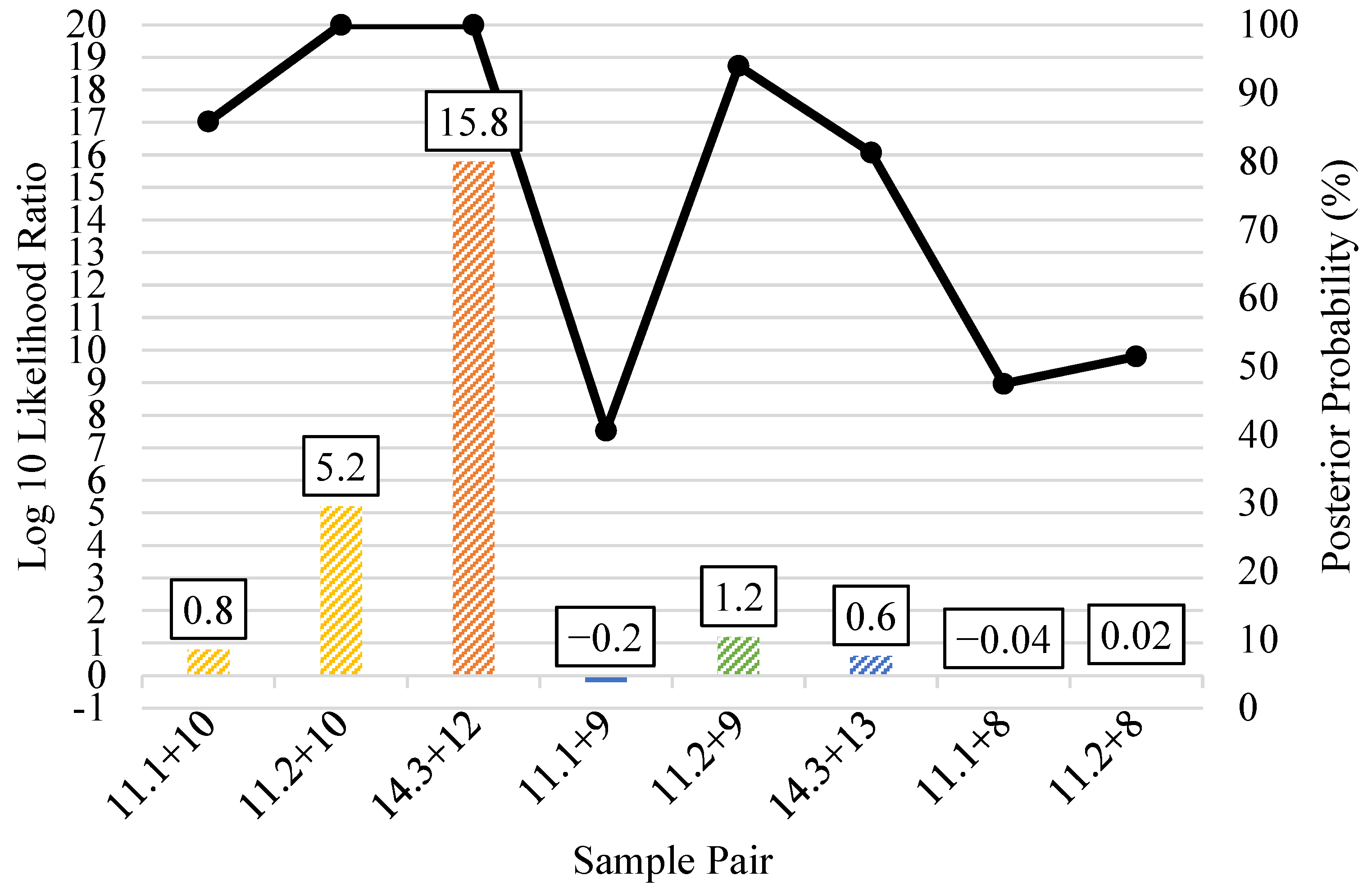
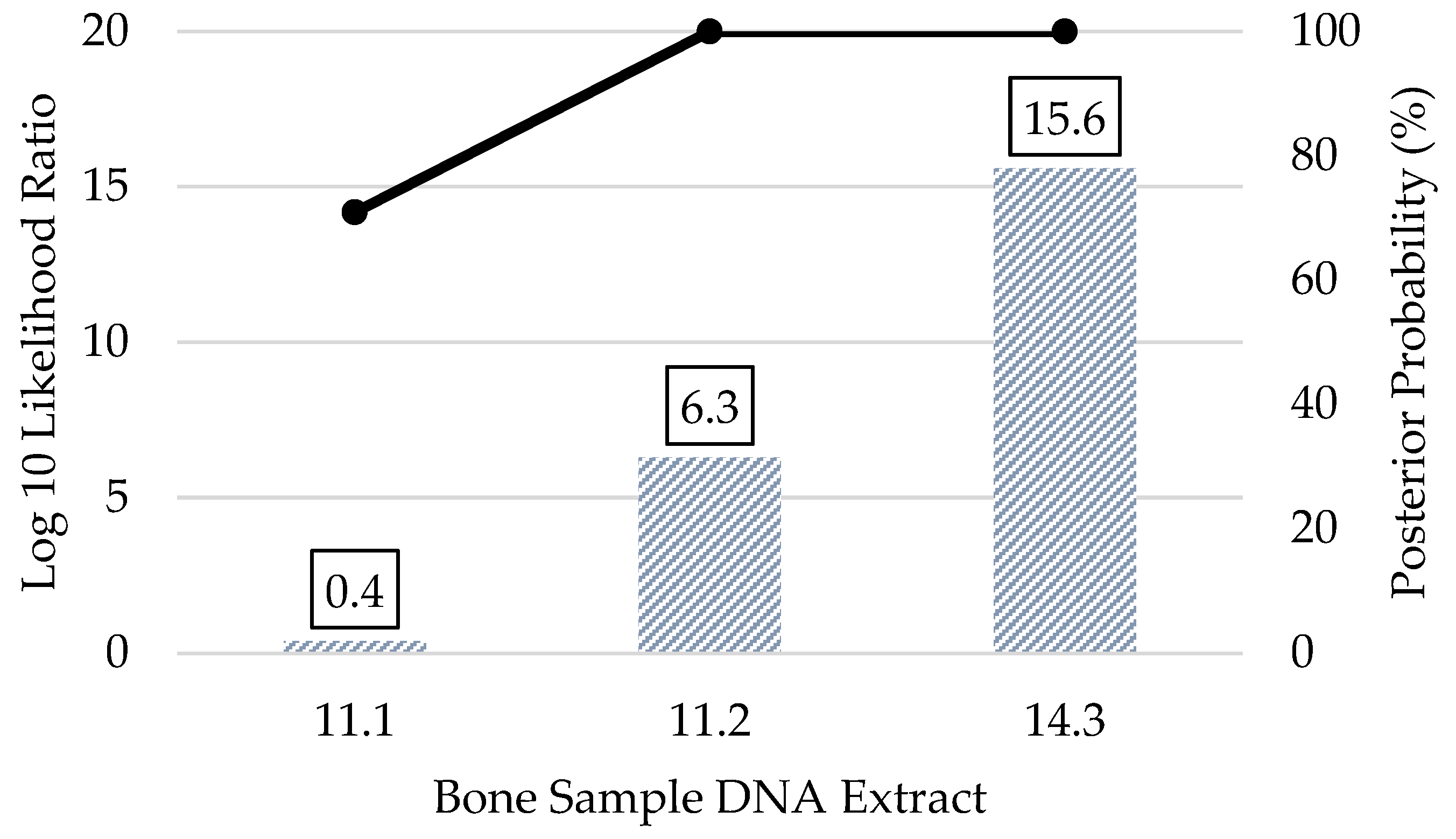
3.5. Kinship Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Butler, J.M.; Hill, C.R. Biology and Genetics of New Autosomal STR Loci Useful for Forensic DNA Analysis. Forensic Sci. Rev. 2012, 24, 15–26. [Google Scholar]
- Kling, D.; Phillips, C.; Kennett, D.; Tillmar, A. Investigative genetic genealogy: Current methods, knowledge and practice. Forensic Sci. Int. Genet. 2021, 52, 102474. [Google Scholar] [CrossRef] [PubMed]
- Erlich, Y.; Shor, T.; Pe’er, I.; Carmi, S. Identity inference of genomic data using long-range familial searches. Science 2018, 362, 690–694. [Google Scholar] [CrossRef] [Green Version]
- GEDmatch. Available online: https://pro.gedmatch.com/user/login (accessed on 11 November 2021).
- Greytak, E.M.; Moore, C.; Armentrout, S.L. Genetic genealogy for cold case and active investigations. Forensic Sci. Int. 2019, 299, 103–113. [Google Scholar] [CrossRef]
- Tillmar, A.; Fagerholm, S.A.; Staaf, J.; Sjolund, P.; Ansell, R. Getting the conclusive lead with investigative genetic genealogy—A successful case study of a 16 year old double murder in Sweden. Forensic Sci. Int. Genet. 2021, 53, 102525. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, D.; Battaglia, J.; Halsing, M.; Sheehan, S. Mitochondrial Sequencing of Missing Persons DNA Casework by Implementing Thermo Fisher’s Precision ID mtDNA Whole Genome Assay. Genes 2020, 11, 1303. [Google Scholar] [CrossRef]
- Holt, C.L.; Stephens, K.M.; Walichiewicz, P.; Fleming, K.D.; Forouzmand, E.; Wu, S.F. Human Mitochondrial Control Region and mtGenome: Design and Forensic Validation of NGS Multiplexes, Sequencing and Analytical Software. Genes 2021, 12, 599. [Google Scholar] [CrossRef] [PubMed]
- Eduardoff, M.; Xavier, C.; Strobl, C.; Casas-Vargas, A.; Parson, W. Optimized mtDNA Control Region Primer Extension Capture Analysis for Forensically Relevant Samples and Highly Compromised mtDNA of Different Age and Origin. Genes 2017, 8, 237. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Oliver, R.S.; Barritt-Ross, S.; McMahon, T.P. Performance evaluation of a mitogenome capture and Illumina sequencing protocol using non-probative, case-type skeletal samples: Implications for the use of a positive control in a next-generation sequencing procedure. Forensic Sci. Int. Genet. 2017, 31, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Bose, N.; Carlberg, K.; Sensabaugh, G.; Erlich, H.; Calloway, C. Target capture enrichment of nuclear SNP markers for massively parallel sequencing of degraded and mixed samples. Forensic Sci. Int. Genet. 2018, 34, 186–196. [Google Scholar] [CrossRef]
- Daniels-Higginbotham, J.; Gorden, E.M.; Farmer, S.K.; Spatola, B.; Damann, F.; Bellantoni, N.; Gagnon, K.S.; de la Puente, M.; Xavier, C.; Walsh, S.; et al. DNA Testing Reveals the Putative Identity of JB55, a 19th Century Vampire Buried in Griswold, Connecticut. Genes 2019, 10, 636. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.; Sturk-Andreaggi, K.; Gorden, E.M.; Daniels-Higginbotham, J.; Sanchez, S.G.; Basic, Z.; Kruzic, I.; Andelinovic, S.; Bosnar, A.; Coklo, M.; et al. A Forensic Genomics Approach for the Identification of Sister Marija Crucifiksa Kozulic. Genes 2020, 11, 938. [Google Scholar] [CrossRef]
- Gorden, E.M.; Greytak, E.M.; Sturk-Andreaggi, K.; Cady, J.; McMahon, T.P.; Armentrout, S.; Marshall, C. Ex-tended kinship analysis of historical remains using SNP capture. Forensic Sci. Int. Genet. 2021, 57, 102636. [Google Scholar] [CrossRef]
- Turchi, C.; Previdere, C.; Bini, C.; Carnevali, E.; Grignani, P.; Manfredi, A.; Melchionda, F.; Onofri, V.; Pelotti, S.; Robino, C.; et al. Assessment of the Precision ID Identity Panel kit on challenging forensic samples. Forensic Sci. Int. Genet. 2020, 49, 102400. [Google Scholar] [CrossRef] [PubMed]
- Ralf, A.; van Oven, M.; Montiel Gonzalez, D.; de Knijff, P.; van der Beek, K.; Wootton, S.; Lagace, R.; Kayser, M. Forensic Y-SNP analysis beyond SNaPshot: High-resolution Y-chromosomal haplogrouping from low quality and quantity DNA using Ion AmpliSeq and targeted massively parallel sequencing. Forensic Sci. Int. Genet. 2019, 41, 93–106. [Google Scholar] [CrossRef]
- Tillmar, A.O.; Kling, D.; Butler, J.M.; Parson, W.; Prinz, M.; Schneider, P.M.; Egeland, T.; Gusmao, L. DNA Commission of the International Society for Forensic Genetics (ISFG): Guidelines on the use of X-STRs in kinship analysis. Forensic Sci. Int. Genet. 2017, 29, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Grandell, I.; Samara, R.; Tillmar, A.O. A SNP panel for identity and kinship testing using massive parallel sequencing. Int. J. Legal Med. 2016, 130, 905–914. [Google Scholar] [CrossRef]
- Xavier, C.; de la Puente, M.; Mosquera-Miguel, A.; Freire-Aradas, A.; Kalamara, V.; Vidaki, A.; Gross, T.E.; Revoir, A.; Pospiech, E.; Kartasinska, E.; et al. Development and validation of the VISAGE AmpliSeq basic tool to predict appearance and ancestry from DNA. Forensic Sci. Int. Genet. 2020, 48, 102336. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef]
- Matise, T.C.; Chen, F.; Chen, W.; De La Vega, F.M.; Hansen, M.; He, C.; Hyland, F.C.; Kennedy, G.C.; Kong, X.; Murray, S.S.; et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007, 17, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Edson, S.M. Extraction of DNA from Skeletonized Postcranial Remains: A Discussion of Protocols and Testing Modalities. J. Forensic Sci. 2019, 64, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Glocke, I.; Aximu-Petri, A.; Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc. 2018, 13, 2447–2461. [Google Scholar] [CrossRef] [PubMed]
- ForenSeq DNA Signature Prep Reference Guide, Rev C; Verogen: San Diego, CA, USA, 2020.
- Zhou, B.; Ho, S.S.; Greer, S.U.; Zhu, X.; Bell, J.M.; Arthur, J.G.; Spies, N.; Zhang, X.; Byeon, S.; Pattni, R.; et al. Comprehensive, integrated, and phased whole-genome analysis of the primary ENCODE cell line K562. Genome Res. 2019, 29, 472–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illumina Platinum Genomes. Available online: https://www.illumina.com/platinumgenomes.html (accessed on 13 November 2021).
- Eberle, M.A.; Fritzilas, E.; Krusche, P.; Kallberg, M.; Moore, B.L.; Bekritsky, M.A.; Iqbal, Z.; Chuang, H.Y.; Humphray, S.J.; Halpern, A.L.; et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017, 27, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prcomp. Available online: https://stat.ethz.ch/R-manual/R-devel/library/stats/html/prcomp.html (accessed on 13 November 2021).
- Santos, C.; Phillips, C.; Gomez-Tato, A.; Alvarez-Dios, J.; Carracedo, A.; Lareu, M.V. Inference of Ancestry in Forensic Analysis II: Analysis of Genetic Data. Methods Mol. Biol. 2016, 1420, 255–285. [Google Scholar] [CrossRef]
- de la Puente, M.; Ruiz-Ramirez, J.; Ambroa-Conde, A.; Xavier, C.; Pardo-Seco, J.; Alvarez-Dios, J.; Freire-Aradas, A.; Mosquera-Miguel, A.; Gross, T.E.; Cheung, E.Y.Y.; et al. Development and Evaluation of the Ancestry Informative Marker Panel of the VISAGE Basic Tool. Genes 2021, 12, 1284. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, L.; Breslin, K.; Zuniga, S.; Wirken, L.; Pospiech, E.; Kukla-Bartoszek, M.; Sijen, T.; Knijff, P.; Liu, F.; Branicki, W.; et al. The HIrisPlex-S system for eye, hair and skin colour prediction from DNA: Introduction and forensic developmental validation. Forensic Sci. Int. Genet. 2018, 35, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Hpsconvertsonline. Available online: https://walshlab.sitehost.iu.edu/hpsconvertsonline.R (accessed on 13 November 2021).
- HirisPlex-S System Webtool. Available online: https://hirisplex.erasmusmc.nl/ (accessed on 13 November 2021).
- Ralf, A.; Gonzalez, D.M.; Zhong, K.; Kayser, M. Yleaf: Software for Human Y-Chromosomal Haplogroup Inference from Next-Generation Sequencing Data. Mol. Biol. Evol. 2018, 35, 1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yleaf Position File. Available online: https://github.com/genid/Yleaf/blob/master/Position_files/WGS_hg38.txt (accessed on 13 November 2021).
- Dimery, I.W.; Ross, D.D.; Testa, J.R.; Gupta, S.K.; Felsted, R.L.; Pollak, A.; Bachur, N.R. Variation amongst K562 cell cultures. Exp. Hematol. 1983, 11, 601–610. [Google Scholar] [PubMed]
- SWGDAM. Recommendations of the SWGDAM ad hoc Working Group on Genotyping Results Reported as Likelihood Ratios; Federal Bureau of Investigation’s Scientific Working Group on DNA Analysis Methods (SWGDAM): Quantico, VA, USA, 2018. [Google Scholar]

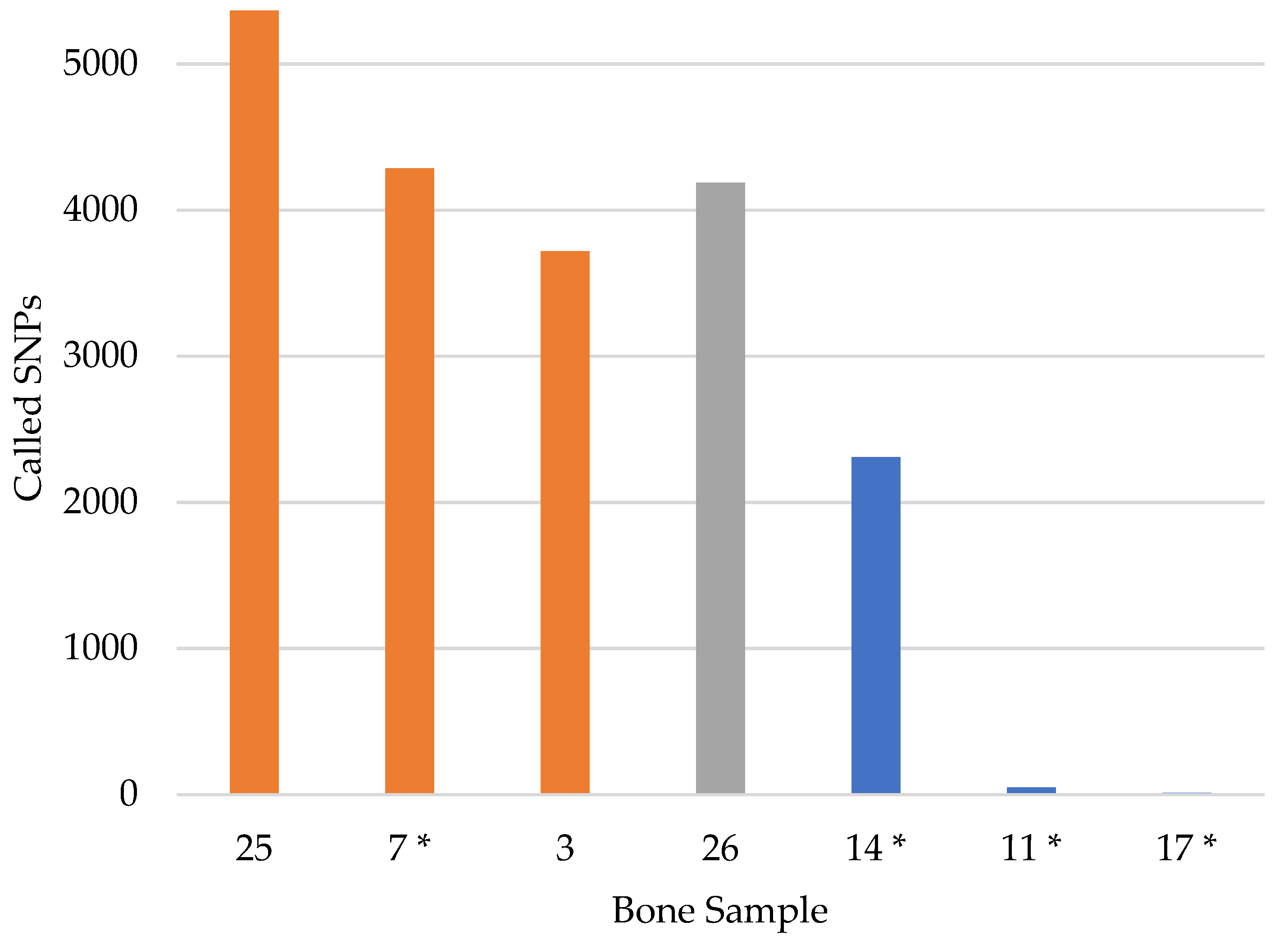


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinship Panel Criterion | Number of Remaining SNPs |
|---|---|
| 1. Included on all major SNP array chips (Table 2) | 116,544 |
| 2. MAF 0.2–0.8 in 1000 Genomes populations | 34,851 |
| 3. Minimum 0.5 cm distance | 5426 |
| 4. LD metric r2 < 0.1 | 5376 |
| 5. Maximum 35% frequency difference between 1000 Genomes populations | 3937 |
| Marker Type | SNP Panels (# SNPs) |
|---|---|
| Kinship SNP/ X-SNP | Infinium Global Screening Array (654,027), Infinium Omni Express (710,000), Infinium CytoSNP-850K (850,000) |
| iiSNP | ForenSeq DNA Signature Prep Kit: Primer Mix A (94) |
| Precision ID Identity Panel (90) | |
| QIAseq Investigator 140 SNP panel (140) | |
| aiSNP | ForenSeq DNA Signature Prep Kit: Primer Mix B (56) |
| Precision ID Ancestry Panel (165) | |
| VISAGE panel (115) | |
| piSNP | ForenSeq DNA Signature Prep Kit: Primer Mix B (24) |
| VISAGE panel (41) | |
| Y-SNP | Precision ID Identity Panel (34) |
| AmpliSeq (884) |
| Case | WWII Context | Sample | Sample Type | Relationship to Service Member (DOR) |
|---|---|---|---|---|
| A | USS Oklahoma | 1 | Buccal swab | Grandniece (3) |
| 2 | Buccal swab | Nephew (2) | ||
| 3 | Left femur | Self | ||
| B | USS Oklahoma | 4 | Buccal swab | Nephew (2) |
| 5 | Buccal swab | Great grandnephew (4) | ||
| 6 | Buccal swab | Great grandniece (4) | ||
| 7.1 | Left femur | Self | ||
| 7.2 | ||||
| C | Austria | 8 | Buccal swab | First cousin twice removed, male (5) |
| 9 | Buccal swab | Nephew (2) | ||
| 10 | Buccal swab | Daughter (1) | ||
| 11.1 | Long bone | Self | ||
| 11.2 | ||||
| D | Italy | 12 | Buccal swab | Sister (1) |
| 13 | Buccal swab | Grandniece (3) | ||
| 14.1 | Right parietal | Self | ||
| 14.2 | ||||
| 14.3 | ||||
| E | Tarawa | 15 | Buccal swab | Son (1) |
| 16 | Buccal swab | Daughter (1) | ||
| 17.1 | Right tibia | Self | ||
| 17.2 |
| Sample | Sample Type | Sex |
|---|---|---|
| 18 | Buccal swab | Female |
| 19 | Buccal swab | Female |
| 20 | Buccal swab | Male |
| 21 | Buccal swab | Female |
| 22 | Buccal swab | Female |
| 23 | Buccal swab | Male |
| 24 | Buccal swab | Male |
| 25 (JB55) | Femoral bone (~200 years) | Male |
| 26 | Unknown bone (~200 years) | Unknown |
| 2800M | Whole blood | Male |
| K562 | Cell line | Female |
| NA12877 | Cell line | Male |
| NA12878 | Cell line | Female |
| Sample Type | Count | Maximum Possible SNPs | Called SNPs * | |||||
|---|---|---|---|---|---|---|---|---|
| Count | Percentage | |||||||
| Minimum | Maximum | Average | Minimum | Maximum | Average | |||
| Male, high quality | 11 | 5422 | 5133 | 5395 | 5355 | 94.7% | 99.5% | 98.8% |
| Female, high quality | 12 | 4593 | 4396 | 4582 | 4552 | 95.7% | 99.8% | 99.1% |
| Bone (all male) | 12 | 5422 | 11 | 5361 | 2407 | 0.2% | 98.9% | 44.4% |
| Control blank | 7 | 5422 | 0 | 26 | 4 | 0.0% | 0.5% | 0.1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tillmar, A.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Thomas, J.T.; Marshall, C. The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications. Genes 2021, 12, 1968. https://doi.org/10.3390/genes12121968
Tillmar A, Sturk-Andreaggi K, Daniels-Higginbotham J, Thomas JT, Marshall C. The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications. Genes. 2021; 12(12):1968. https://doi.org/10.3390/genes12121968
Chicago/Turabian StyleTillmar, Andreas, Kimberly Sturk-Andreaggi, Jennifer Daniels-Higginbotham, Jacqueline Tyler Thomas, and Charla Marshall. 2021. "The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications" Genes 12, no. 12: 1968. https://doi.org/10.3390/genes12121968
APA StyleTillmar, A., Sturk-Andreaggi, K., Daniels-Higginbotham, J., Thomas, J. T., & Marshall, C. (2021). The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications. Genes, 12(12), 1968. https://doi.org/10.3390/genes12121968