
Genome-Wide Analysis of the MADS-Box Gene Family in Maize: Gene Structure, Evolution, and Relationships



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification and Classification of MADS-Box Proteins in Maize
2.2. Phylogenetic Analysis and Classification of the MADS Gene Family
2.3. Chromosomal Distribution and Gene Structure Analysis of MADS-Box Genes in Zea mays
2.4. Gene Duplication and Synteny Analysis of MADS-Box Genes in Zea mays
2.5. Expression and Regulation Relationships among MADS-Box Gene Family Members in Maize
3. Results
3.1. Identification of MADS-Box Gene Family Members in Maize
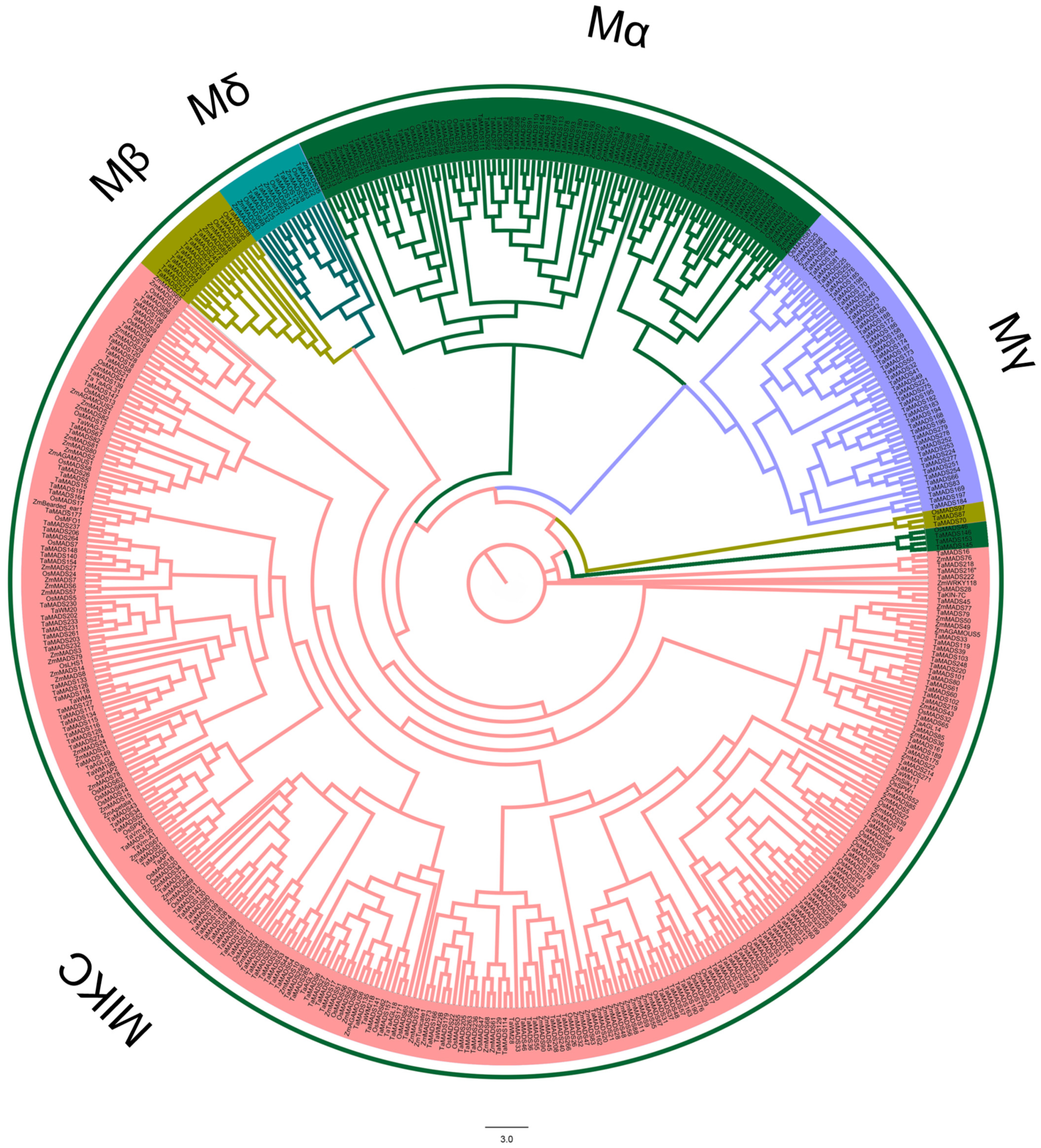
3.2. Classification and Phylogenetic Analysis of the MADS-Box Gene Family Members
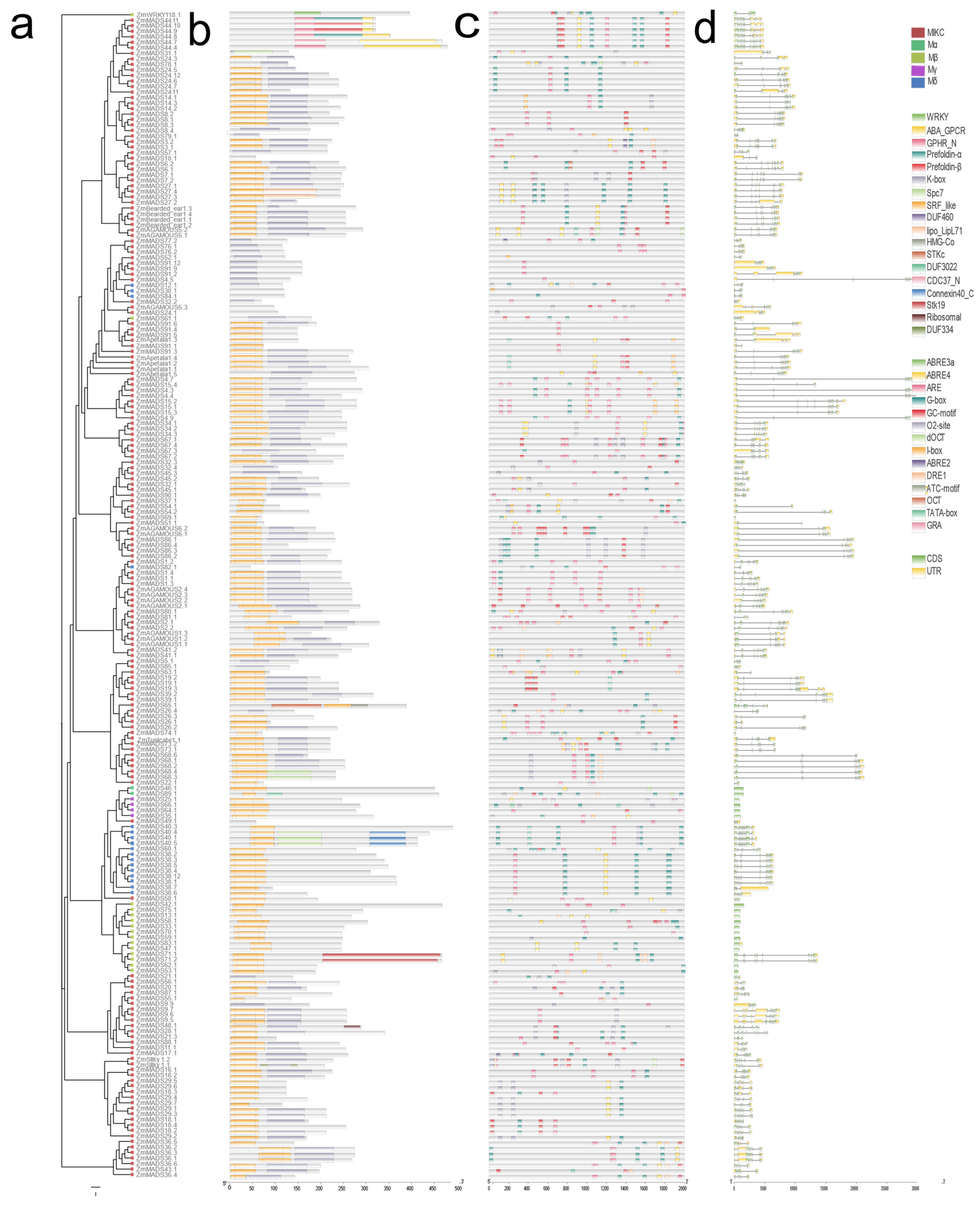
3.3. Gene Structure and Motif Composition of the MADS-Box Gene Family
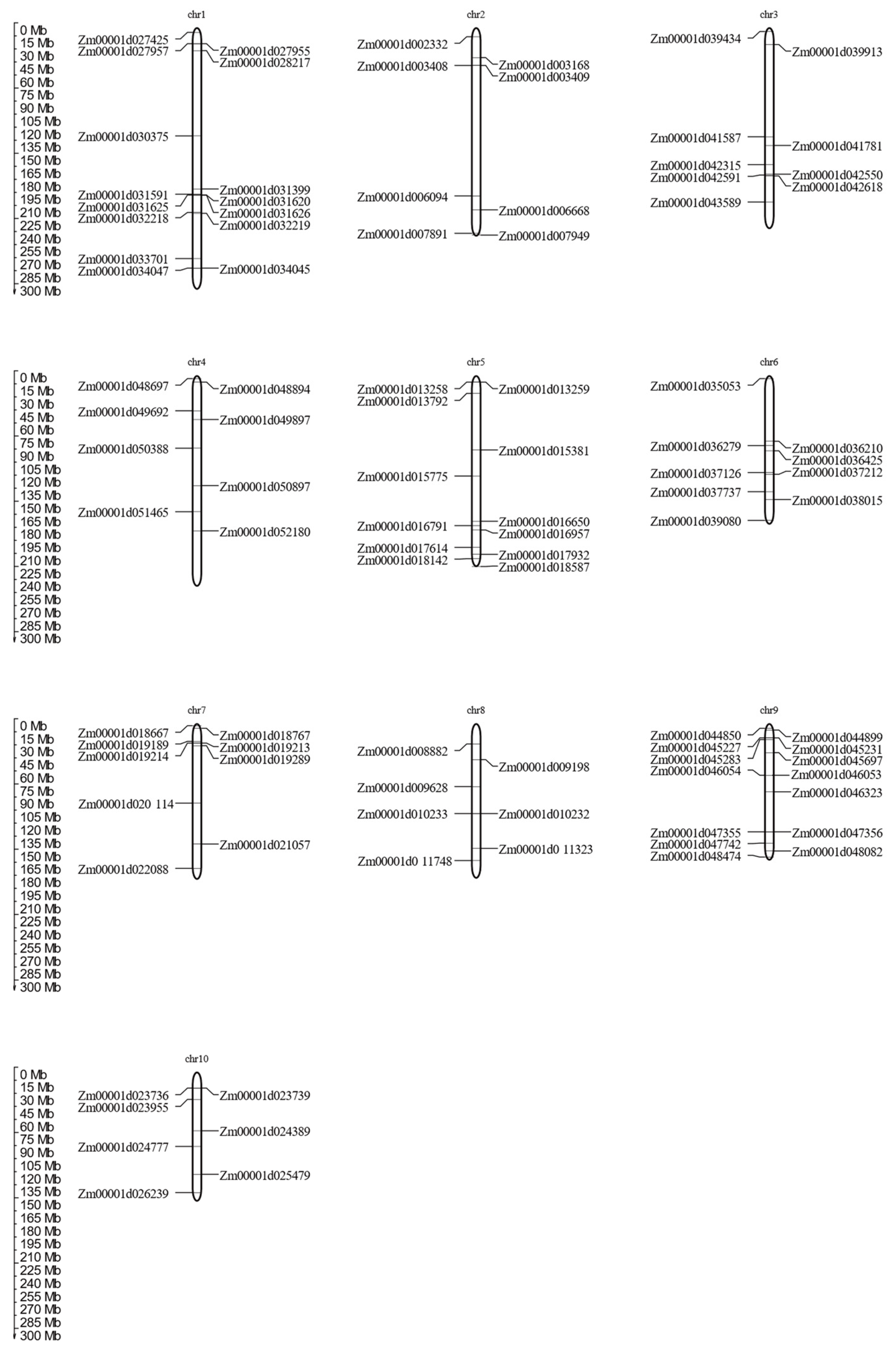
3.4. Gene Dupulacation, Chromosomal Distribution, and Synteny Analysis of MADS-Box Gene Family Members
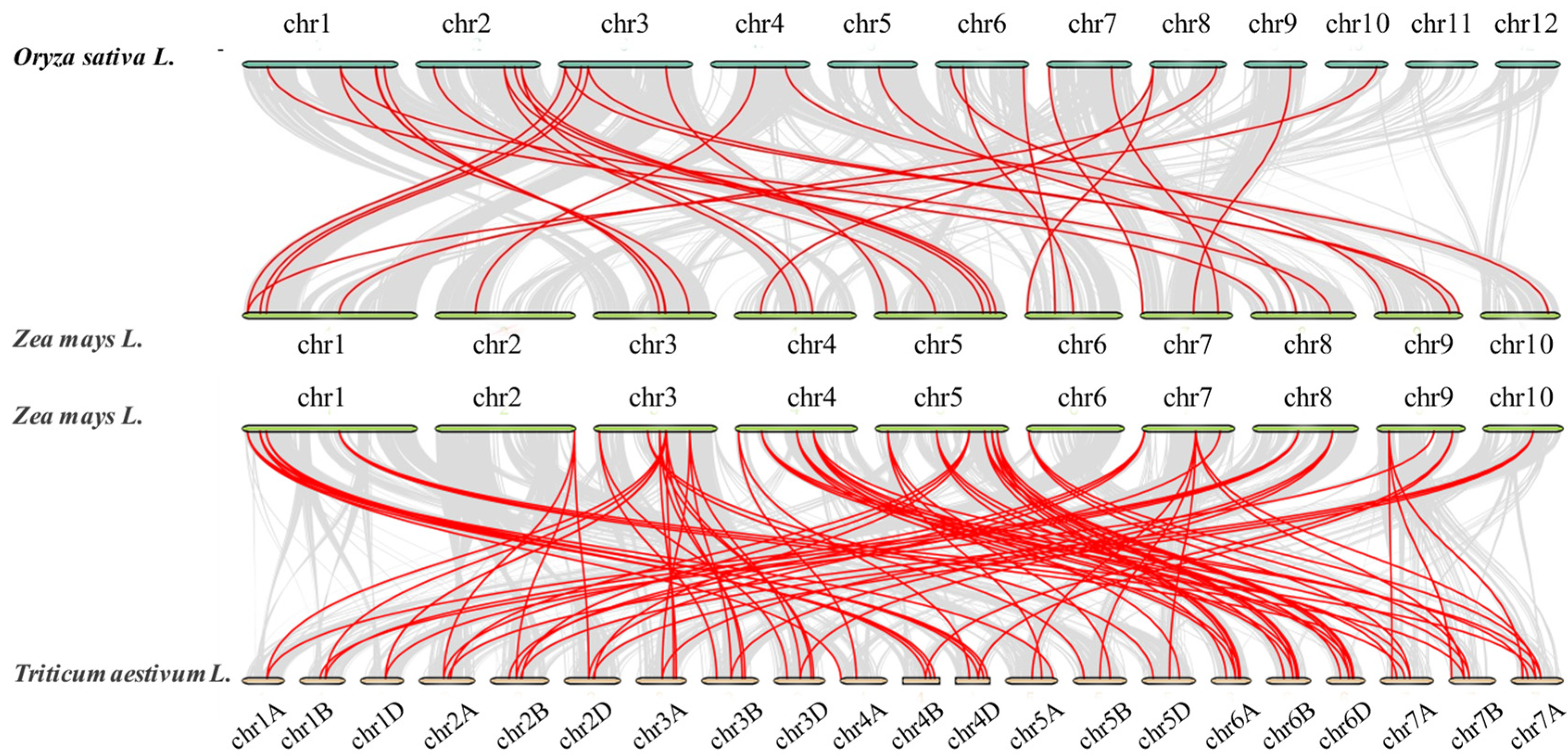
3.5. Synteny Analysis of MADS-Box Gene Family Members in Maize and Other Species
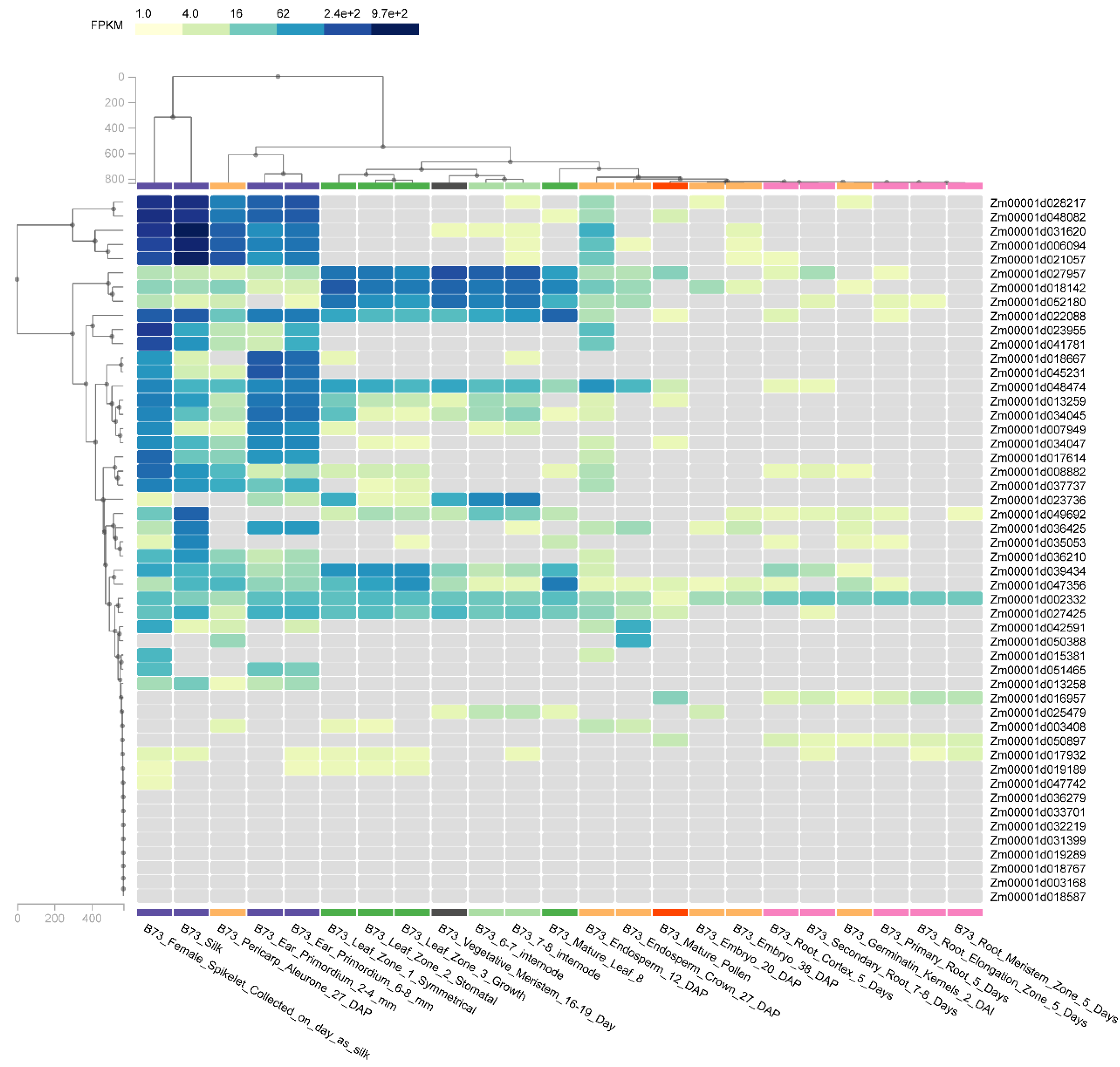
3.6. Gene Expression in MADS-Box Gene Family Members
3.7. Gene Regulation Networks: Review of the Potential Connections
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benz, B.F. Archaeological evidence of teosinte domestication from Guila Naquitz, Oaxaca. Proc. Natl. Acad. Sci. USA 2001, 98, 2104–2106. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. Maize genome mapped. Nature 2009. [Google Scholar] [CrossRef]
- Lawrence, C.J.; Dong, Q.; Polacco, M.L.; Seigfried, T.E.; Brendel, V. MaizeGDB, the community database for maize genetics and genomics. Nucleic Acids Res. 2004, 32, D393–D397. [Google Scholar] [CrossRef] [Green Version]
- Schwarz-Sommer, Z.; Huijser, P.; Nacken, W.; Saedler, H.; Sommer, H. Genetic Control of Flower Development by Homeotic Genes in Antirrhinum majus. Science 1990, 250, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treisman, R. DNA-binding proteins. Inside the MADS box. Nature 1995, 376, 468–469. [Google Scholar] [CrossRef]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Buylla, E.R.; Liljegren, S.J.; Pelaz, S.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Vergara-Silva, F.; Yanofsky, M.F. MADS-box gene evolution beyond flowers: Expression in pollen, endosperm, guard cells, roots and trichomes. Plant J. 2008, 24, 457–466. [Google Scholar] [CrossRef]
- Kaufmann, K.; Melzer, R.; Theissen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef]
- Parenicova, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.; Kim, J.; Lee, S.; An, G.; Ma, H.; Nei, M. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. USA 2004, 101, 1910–1915. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henschel, K.; Kofuji, R.; Hasebe, M.; Saedler, H.; Munster, T.; Theissen, G. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol. Biol. Evol. 2002, 19, 801–814. [Google Scholar] [CrossRef]
- Theißen, G.; Gramzow, L. Structure and Evolution of Plant MADS Domain Transcription Factors. In Plant Transcription Factors; Academic Press: Cambridge, MA, USA, 2016; pp. 127–138. [Google Scholar]
- Mena, M.; Mandel, M.A.; Lerner, D.R.; Yanofsky, M.F.; Schmidt, R.J. A characterization of the MADS-box gene family in maize. Plant J. 1995, 8, 845–854. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, Z.; Li, X.; Wang, Z.; Chen, F.; Mi, G.; Forde, B.; Takahashi, H.; Yuan, L. Involvement of a truncated MADS-box transcription factor ZmTMM1 in root nitrate foraging. J. Exp. Bot. 2020, 71, 4547–4561. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Wang, F.; Kong, J.; Xu, Q.; Li, T.; Chen, L.; Chen, H.; Jiang, H.; Li, C.; Cheng, B. Functional analysis of ZmMADS1a reveals its role in regulating starch biosynthesis in maize endosperm. Sci. Rep. 2019, 9, 3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Lawit, S.J.; Weers, B.; Sun, J.; Mongar, N.; Van Hemert, J.; Melo, R.; Meng, X.; Rupe, M.; Clapp, J.; et al. Overexpression of zmm28 increases maize grain yield in the field. Proc. Natl. Acad. Sci. USA 2019, 116, 23850–23858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Esawi, M.A.; Alayafi, A.A. Overexpression of Rice Rab7 Gene Improves Drought and Heat Tolerance and Increases Grain Yield in Rice (Oryza sativa L.). Genes 2019, 10, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012, 40, D302–D305. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Gui, S.; Yang, L.; Li, J.; Luo, J.; Xu, X.; Yuan, J.; Chen, L.; Li, W.; Yang, X.; Wu, S.; et al. ZEAMAP, a Comprehensive Database Adapted to the Maize Multi-Omics Era. iScience 2020, 23, 101241. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Yang, Y.; Luo, W.; Yang, C.; Ding, P.; Liu, Y.; Qiao, L.; Chang, Z.; Geng, H.; Wang, P.; et al. Genome-wide identification and analysis of the MADS-box gene family in bread wheat (Triticum aestivum L.). PLoS ONE 2017, 12, e0181443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Adams, C.I.M.; Knapp, M.; Gemmell, N.J.; Jeunen, G.J.; Bunce, M.; Lamare, M.D.; Taylor, H.R. Beyond Biodiversity: Can Environmental DNA (eDNA) Cut It as a Population Genetics Tool? Genes 2019, 10, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingan, S.B.; Heaton, H.; Cudini, J.; Lambert, C.C.; Baybayan, P.; Galvin, B.D.; Durbin, R.; Korlach, J.; Lawniczak, M.K.N. A High-Quality De novo Genome Assembly from a Single Mosquito Using PacBio Sequencing. Genes 2019, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Shen, Z.; Zhao, D.; Xu, L.; Zhang, L.; Zou, Q. Genome-Wide Analysis of LysM-Containing Gene Family in Wheat: Structural and Phylogenetic Analysis during Development and Defense. Genes 2020, 12, 31. [Google Scholar] [CrossRef]
- Leseberg, C.H.; Li, A.; Kang, H.; Duvall, M.; Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 2006, 378, 84–94. [Google Scholar] [CrossRef]
- Duan, W.; Song, X.; Liu, T.; Huang, Z.; Ren, J.; Hou, X.; Li, Y. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genom. 2015, 290, 239–255. [Google Scholar] [CrossRef]
- Wei, B.; Zhang, R.Z.; Guo, J.J.; Liu, D.M.; Li, A.L.; Fan, R.C.; Mao, L.; Zhang, X.Q. Genome-wide analysis of the MADS-box gene family in Brachypodium distachyon. PLoS ONE 2014, 9, e84781. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Wang, J.; Bao, X.; Wu, Q.; Yang, T.; Li, H.; Wang, W.; Zhang, Y.; Bai, N.; Guan, Y.; et al. Genome-wide analysis of Jatropha curcas MADS-box gene family and functional characterization of the JcMADS40 gene in transgenic rice. BMC Genom. 2020, 21, 325. [Google Scholar] [CrossRef]
- Zheng, Y.; Ren, N.; Wang, H.; Stromberg, A.J.; Perry, S.E. Global Identification of Targets of the Arabidopsis MADS Domain Protein AGAMOUS-Like15. Plant Cell 2009, 21, 2563–2577. [Google Scholar] [CrossRef] [Green Version]
- Gramzow, L.; Ritz, M.S.; Theissen, G. On the origin of MADS-domain transcription factors. Trends Genet. 2010, 26, 149–153. [Google Scholar] [CrossRef]
- Adamczyk, B.J.; Fernandez, D.E. MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 2009, 149, 1713–1723. [Google Scholar] [CrossRef] [Green Version]
- Motorin, Y.; Helm, M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes 2019, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Schilling, S.; Pan, S.; Kennedy, A.; Melzer, R. MADS-box genes and crop domestication: The jack of all traits. J. Exp. Bot. 2018, 69, 1447–1469. [Google Scholar] [CrossRef]
- Haines, B.E.; Steussy, C.N.; Stauffacher, C.V.; Wiest, O. Molecular modeling of the reaction pathway and hydride transfer reactions of HMG-CoA reductase. Biochemistry 2012, 51, 7983–7995. [Google Scholar] [CrossRef] [Green Version]
- Rodwell, V.W.; Nordstrom, J.L.; Mitschelen, J.J. Regulation of HMG-CoA reductase. Adv. Lipid Res. 1976, 14, 1–74. [Google Scholar] [CrossRef]
- Pandey, S.; Nelson, D.C.; Assmann, S.M. Two Novel GPCR-Type G Proteins Are Abscisic Acid Receptors in Arabidopsis. Cell 2009, 136, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Sahlan, M.; Zako, T.; Yohda, M. Prefoldin, a jellyfish-like molecular chaperone: Functional cooperation with a group II chaperonin and beyond. Biophys. Rev. 2018, 10, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Kerres, A.; Vietmeier-Decker, C.; Ortiz, J.; Karig, I.; Beuter, C.; Hegemann, J.; Lechner, J.; Fleig, U. The fission yeast kinetochore component Spc7 associates with the EB1 family member Mal3 and is required for kinetochore-spindle association. Mol. Biol. Cell 2004, 15, 5255–5267. [Google Scholar] [CrossRef] [Green Version]
- Terzaghi, W.B.; Cashmore, A.R. Light-Regulated Transcription. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1995, 46, 445–474. [Google Scholar] [CrossRef]
- Yoshida, T.; Fujita, Y.; Sayama, H.; Kidokoro, S.; Maruyama, K.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AREB1, AREB2, and ABF3 are master transcription factors that cooperatively regulate ABRE-dependent ABA signaling involved in drought stress tolerance and require ABA for full activation. Plant J. 2010, 61, 672–685. [Google Scholar] [CrossRef]
- Fujita, Y.; Yoshida, T.; Yamaguchi-Shinozaki, K. Pivotal role of the AREB/ABF-SnRK2 pathway in ABRE-mediated transcription in response to osmotic stress in plants. Physiol. Plant 2013, 147, 15–27. [Google Scholar] [CrossRef]
- Arora, K.; Panda, K.K.; Mittal, S.; Mallikarjuna, M.G.; Thirunavukkarasu, N. In Silico Characterization and Functional Validation of Cell Wall Modification Genes Imparting Waterlogging Tolerance in Maize. Bioinform. Biol. Insights 2017, 11, 1177932217747277. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, G.; da Silva, J.A.T.; Zhao, C.; Duan, J. The methyl jasmonate-responsive transcription factor DobHLH4 promotes DoTPS10, which is involved in linalool biosynthesis in Dendrobium officinale during floral development. Plant Sci. 2021, 309, 110952. [Google Scholar] [CrossRef]
- Shu, Y.; Yu, D.; Wang, D.; Guo, D.; Guo, C. Genome-wide survey and expression analysis of the MADS-box gene family in soybean. Mol. Biol. Rep. 2013, 40, 3901–3911. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Fu, Q.; Ma, Z.; Sun, W.; Huang, L.; Wu, Q.; Tang, Z.; Bu, T.; Li, C.; Chen, H. Genome-wide investigation of the MADS gene family and dehulling genes in tartary buckwheat (Fagopyrum tataricum). Planta 2019, 249, 1301–1318. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, X.; Chen, W.; Peng, X.; Cheng, X.; Zhu, S.; Cheng, B. Whole-genome survey and characterization of MADS-box gene family in maize and sorghum. Plant Cell Tissue Organ Cult. (PCTOC) 2010, 105, 159–173. [Google Scholar] [CrossRef]
- Ouyang, S.; Zhu, W.; Hamilton, J.; Lin, H.; Campbell, M.; Childs, K.; Thibaud-Nissen, F.; Malek, R.L.; Lee, Y.; Zheng, L.; et al. The TIGR Rice Genome Annotation Resource: Improvements and new features. Nucleic Acids Res. 2007, 35, D883–D887. [Google Scholar] [CrossRef] [Green Version]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361. [Google Scholar] [CrossRef] [Green Version]
- Ohta, T. Gene Families: Multigene Families and Superfamilies. In Encyclopedia of Life Sciences; ELS: Chichester, UK, 2008. [Google Scholar]
- Ohta, T. Gene conversion and evolution of gene families: An overview. Genes 2010, 1, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Xu, R.; Luo, X.; Jiang, Z.; Shu, H. Genome-wide identification and expression analysis of MAPK and MAPKK gene family in Malus domestica. Gene 2013, 531, 377–387. [Google Scholar] [CrossRef]
- Mendes-Moreira, P.; Alves, M.L.; Satovic, Z.; Dos Santos, J.P.; Santos, J.N.; Souza, J.C.; Pego, S.E.; Hallauer, A.R.; Vaz Patto, M.C. Genetic Architecture of Ear Fasciation in Maize (Zea mays) under QTL Scrutiny. PLoS ONE 2015, 10, e0124543. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.E.; Bartling, L.; Whipple, C.; Hall, D.H.; Sakai, H.; Schmidt, R.; Hake, S. bearded-ear encodes a MADS box transcription factor critical for maize floral development. Plant Cell 2009, 21, 2578–2590. [Google Scholar] [CrossRef] [Green Version]
- Bowman, J.L.; Alvarez, J.; Weigel, D.; Meyerowitz, E.M.; Smyth, D.R. Control of flower development in Arabidopsis thaliana by APETALA1 and interacting genes. Development 1993, 119, 721–743. [Google Scholar] [CrossRef]
- Mena, M.; Ambrose, B.A.; Meeley, R.B.; Briggs, S.P.; Yanofsky, M.F.; Schmidt, R.J. Diversification of C-function activity in maize flower development. Science 1996, 274, 1537–1540. [Google Scholar] [CrossRef]
- Laudencia-Chingcuanco, D.; Hake, S. The indeterminate floral apex1 gene regulates meristem determinacy and identity in the maize inflorescence. Development 2002, 129, 2629–2638. [Google Scholar] [CrossRef]
- Han, J.J.; Jackson, D.; Martienssen, R. Pod corn is caused by rearrangement at the Tunicate1 locus. Plant Cell 2012, 24, 2733–2744. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.N. Hybrids of Zea Tunicata and Zea Ramosa. Proc. Natl. Acad. Sci. USA 1917, 3, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Langdale, J.A.; Irish, E.E.; Nelson, T.M. Action of the Tunicate locus on maize floral development. Dev. Genet. 1994, 15, 176–187. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.; Chen, Z.; Xu, L.; Zhang, L.; Zou, Q. Genome-Wide Analysis of the MADS-Box Gene Family in Maize: Gene Structure, Evolution, and Relationships. Genes 2021, 12, 1956. https://doi.org/10.3390/genes12121956
Zhao D, Chen Z, Xu L, Zhang L, Zou Q. Genome-Wide Analysis of the MADS-Box Gene Family in Maize: Gene Structure, Evolution, and Relationships. Genes. 2021; 12(12):1956. https://doi.org/10.3390/genes12121956
Chicago/Turabian StyleZhao, Da, Zheng Chen, Lei Xu, Lijun Zhang, and Quan Zou. 2021. "Genome-Wide Analysis of the MADS-Box Gene Family in Maize: Gene Structure, Evolution, and Relationships" Genes 12, no. 12: 1956. https://doi.org/10.3390/genes12121956
APA StyleZhao, D., Chen, Z., Xu, L., Zhang, L., & Zou, Q. (2021). Genome-Wide Analysis of the MADS-Box Gene Family in Maize: Gene Structure, Evolution, and Relationships. Genes, 12(12), 1956. https://doi.org/10.3390/genes12121956