
Juvenile Amyotrophic Lateral Sclerosis: A Review


Abstract
:1. Introduction
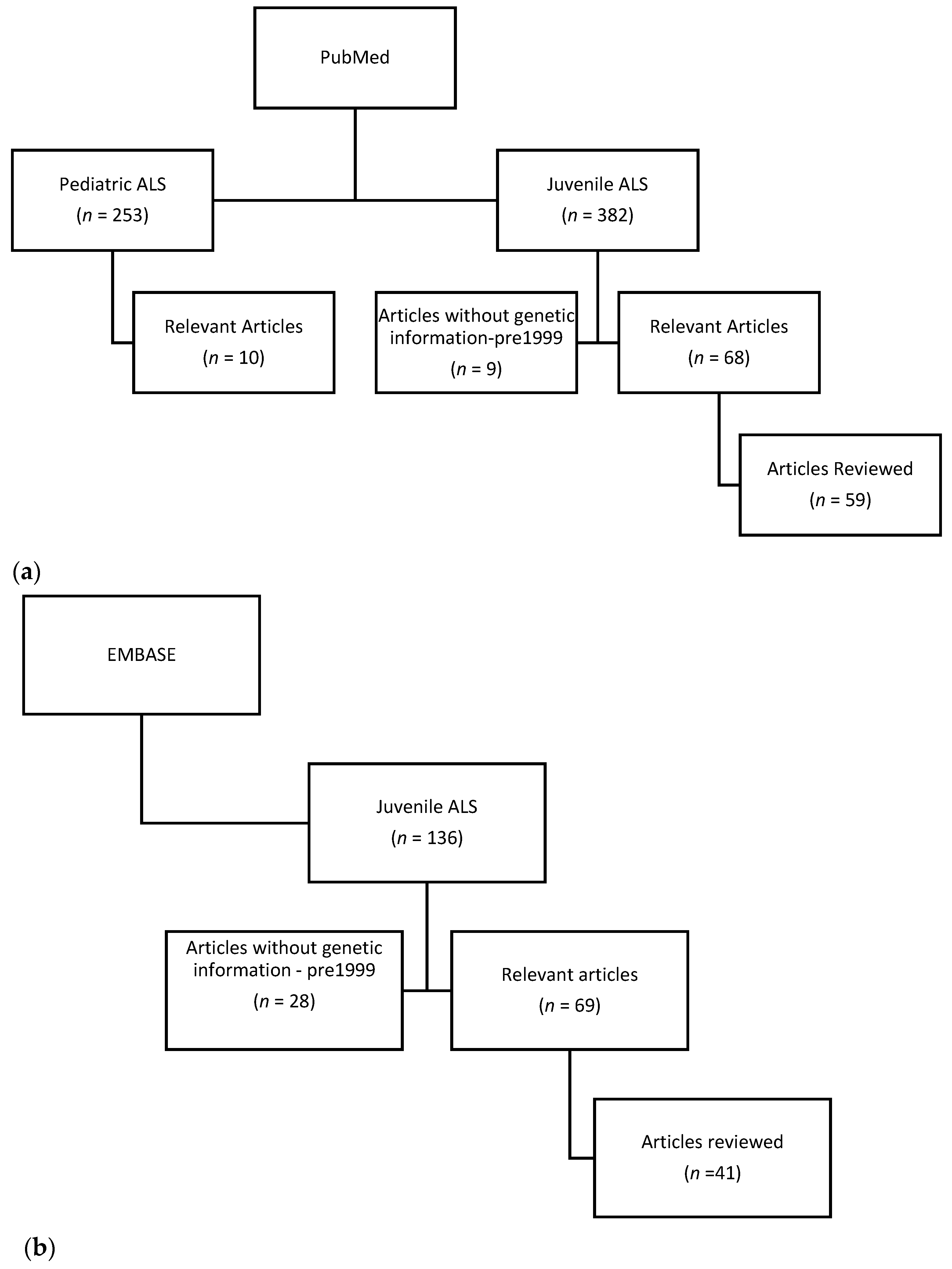
2. Materials and Methods
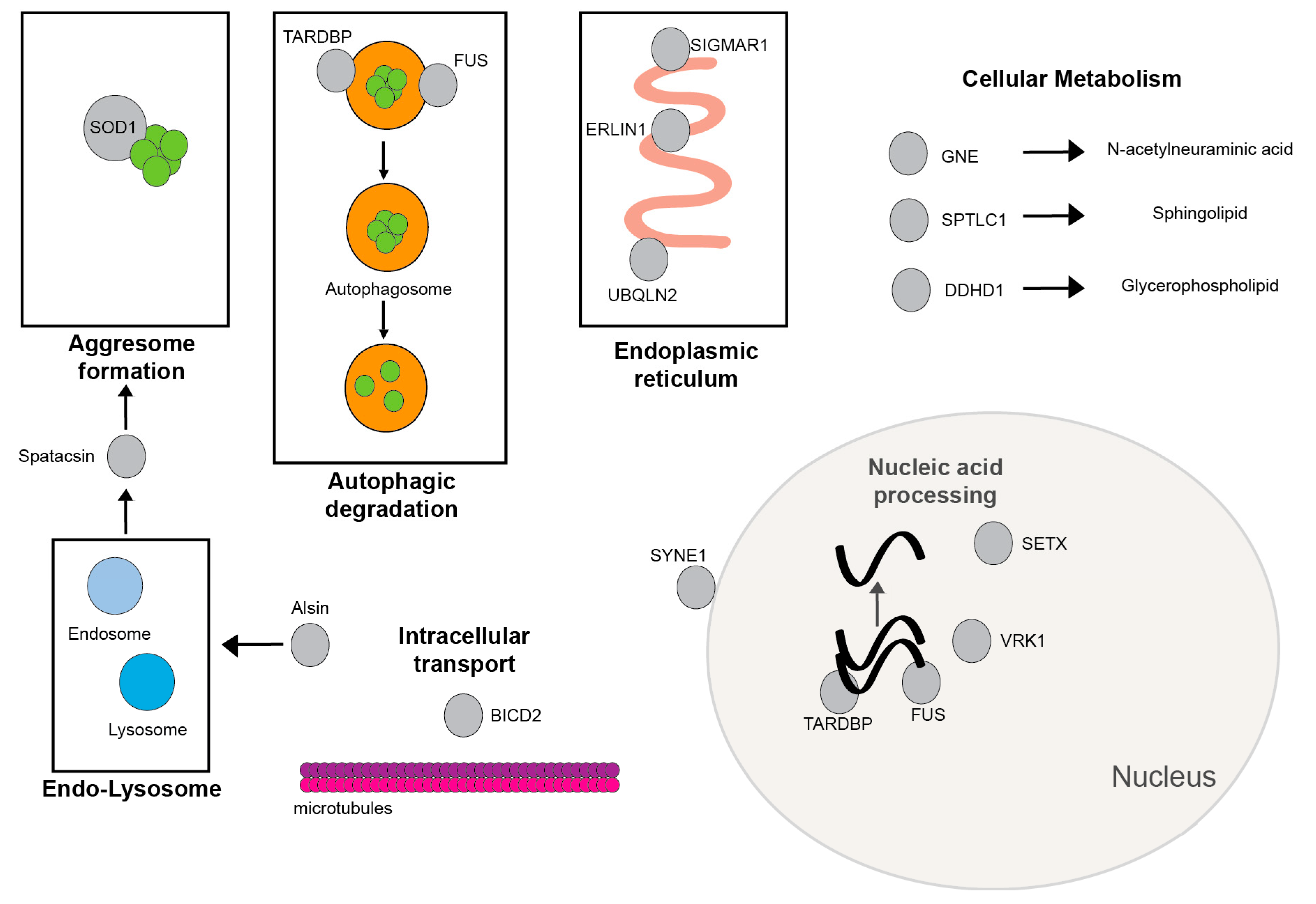
3. Results
3.1. Most Common Gene Associations with JALS
3.1.1. FUS
3.1.2. SETX
3.1.3. ALS2/Alsin
3.2. Rarer Gene Associations with JALS
3.2.1. SIGMAR1
3.2.2. SOD1
3.2.3. SPTLC1
3.2.4. Spatacsin (SPG11)
3.2.5. UBQLN2
3.2.6. ERLIN1
3.2.7. GNE
3.2.8. TARDBP
3.2.9. VRK1
3.2.10. SYNE1
3.2.11. BICD2
3.2.12. DDHD1
3.3. JALS Mimics
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| ALS2 | amyotrophic lateral sclerosis 2 |
| ALS4 | amyotrophic lateral sclerosis 4 |
| AO-ALS | adult onset amyotrophic lateral sclerosis |
| BICD2 | BICD cargo adaptor 2 gene |
| DDHD1 | DDHD domain containing 1 |
| dHMN | distal hereditary motor neuropathy |
| EMG | electromyography |
| ERLIN1 | endoplasmic reticulum lipid raft-associated protein 1 |
| fMRI | functional magnetic resonance imaging |
| FTD | frontotemporal dementia |
| FUS | fused in Sarcoma |
| GNE | glucosamine (UDP-N-acetyl)-2-epimerase |
| HSAN | hereditary sensory autonomic neuropathy |
| HSP | hereditary spastic paraplegia |
| JALS | juvenile amyotrophic lateral sclerosis |
| JPLS | juvenile primary lateral sclerosis |
| LMN | lower motor neuron |
| MRI | magnetic resonance imaging |
| SIGMAR1 | Sigma-1 receptor |
| SMA | spinal muscular atrophy |
| SOD1 | copper/zinc superoxide dismutase-1 |
| SPG11 | spastic paraplegia 11 |
| SPTLC1 | serine palmitoyltransferase, long-chain base subunit 1 |
| SYNE1 | spectrin repeat containing nuclear envelope protein 1 gene |
| TARDBP | TAR DNA binding protein |
| TDP 43 | transactive response DNA binding protein 43 |
| UBQNL2 | Ubiquilin2 |
| UMN | upper motor neuron |
| VRK1 | vaccinia-related kinase 1 |
References
- Liu, Z.J.; Lin, H.X.; Liu, G.L.; Tao, Q.Q.; Ni, W.; Xiao, B.G.; Wu, Z.Y. The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Shashiraj. Juvenile amyotrophic lateral sclerosis. Indian J. Pediatr. 2006, 73, 225–226. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Brunet, F.; Dupre, N.; Chrestian, N. The Occurrence of FUS Mutations in Pediatric Amyotrophic Lateral Sclerosis: A Case Report and Review of the Literature. J. Child. Neurol. 2020, 35, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Kacem, I.; Sghaier, I.; Bougatef, S.; Nasri, A.; Gargouri, A.; Ajroud-Driss, S.; Gouider, R. Epidemiological and clinical features of amyotrophic lateral sclerosis in a Tunisian cohort. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef]
- Hubers, A.; Just, W.; Rosenbohm, A.; Muller, K.; Marroquin, N.; Goebel, I.; Hogel, J.; Thiele, H.; Altmuller, J.; Nurnberg, P.; et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol. Aging 2015, 36, 3117.e3111–3117.e3116. [Google Scholar] [CrossRef]
- Chen, L. FUS mutation is probably the most common pathogenic gene for JALS, especially sporadic JALS. Rev. Neurol. 2021, 177, 333–340. [Google Scholar] [CrossRef]
- Zou, Z.Y.; Che, C.H.; Feng, S.Y.; Fang, X.Y.; Huang, H.P.; Liu, C.Y. Novel FUS mutation Y526F causing rapidly progressive familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2021, 22, 73–79. [Google Scholar] [CrossRef]
- Naumann, M.; Peikert, K.; Gunther, R.; van der Kooi, A.J.; Aronica, E.; Hubers, A.; Danel, V.; Corcia, P.; Pan-Montojo, F.; Cirak, S.; et al. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2384–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirayanagi, K.; Sato, M.; Furuta, N.; Makioka, K.; Ikeda, Y. Juvenile-onset Sporadic Amyotrophic Lateral Sclerosis with a Frameshift FUS Gene Mutation Presenting Unique Neuroradiological Findings and Cognitive Impairment. Intern. Med. 2016, 55, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, J.; Lu, H.; Liu, Y. A de novo c.1509dupA:p.R503fs mutation of FUS: Report of a girl with sporadic juvenile amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2020, 21, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.C.; Power, R.; Ealing, J.; Hamdalla, H. FUS-ALS presenting with myoclonic jerks in a 17-year-old man. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2019, 20, 278–280. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, H.; Cai, Y.; Wen, H.; Wang, L.; Zhu, M.; Chen, Y.; Yu, Y.; Lu, X.; Zhou, M.; et al. FUS P525L mutation causing amyotrophic lateral sclerosis and movement disorders. Brain Behav. 2020, 10, e01625. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Potocki, L.; Collier, T.R.; Woodbury, S.L.; Adesina, A.M.; Jones, J.; Lotze, T.E. Utility of whole exome sequencing in evaluation of juvenile motor neuron disease. Muscle Nerve. 2016, 53, 648–652. [Google Scholar] [CrossRef]
- Leblond, C.S.; Webber, A.; Gan-Or, Z.; Moore, F.; Dagher, A.; Dion, P.A.; Rouleau, G.A. De novo FUS P525L mutation in Juvenile amyotrophic lateral sclerosis with dysphonia and diplopia. Neurol. Genet. 2016, 2, e63. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zhao, Z.; Shen, H.; Bing, Q.; Li, N.; Hu, J. Clinical and Genetic Features of Patients with Juvenile Amyotrophic Lateral Sclerosis with Fused in Sarcoma (FUS) Mutation. Med. Sci. Monit. 2018, 24, 8750–8757. [Google Scholar] [CrossRef] [PubMed]
- Baumer, D.; Hilton, D.; Paine, S.M.; Turner, M.R.; Lowe, J.; Talbot, K.; Ansorge, O. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology 2010, 75, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Ansorge, O.; Strong, M.; Bilbao, J.; Zinman, L.; Ang, L.C.; Baker, M.; Stewart, H.; Eisen, A.; Rademakers, R.; et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: Two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011, 122, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscul. Disord. 2012, 22, 73–75. [Google Scholar] [CrossRef]
- Zou, Z.Y.; Liu, M.S.; Li, X.G.; Cui, L.Y. Mutations in SOD1 and FUS caused juvenile-onset sporadic amyotrophic lateral sclerosis with aggressive progression. Ann. Transl. Med. 2015, 3, 221. [Google Scholar] [CrossRef] [PubMed]
- Grunseich, C.; Patankar, A.; Amaya, J.; Watts, J.A.; Li, D.; Ramirez, P.; Schindler, A.B.; Fischbeck, K.H.; Cheung, V.G. Clinical and Molecular Aspects of Senataxin Mutations in Amyotrophic Lateral Sclerosis 4. Ann. Neurol. 2020, 87, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, P.F.; Rabin, B.A.; Ryan, S.G.; Ding, Y.; Scavina, M.; Crain, B.; Griffin, J.W.; Cornblath, D.R. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am. J. Hum. Genet. 1998, 62, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- Rabin, B.A.; Griffin, J.W.; Crain, B.J.; Scavina, M.; Chance, P.F.; Cornblath, D.R. Autosomal dominant juvenile amyotrophic lateral sclerosis. Brain 1999, 122, 1539–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algahtani, H.; Shirah, B.; Algahtani, R.; Naseer, M.I.; Al-Qahtani, M.H.; Abdulkareem, A.A. Ataxia with ocular apraxia type 2 not responding to 4-aminopyridine: A rare mutation in the SETX gene in a Saudi patient. Intractable Rare Dis. Res. 2018, 7, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Nanetti, L.; Cavalieri, S.; Pensato, V.; Erbetta, A.; Pareyson, D.; Panzeri, M.; Zorzi, G.; Antozzi, C.; Moroni, I.; Gellera, C.; et al. SETX mutations are a frequent genetic cause of juvenile and adult onset cerebellar ataxia with neuropathy and elevated serum α-fetoprotein. Orphanet J. Rare Dis. 2013, 8, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, P.; Feng, S.; Tsai, Y.L.; Li, W.; Rinchetti, P.; Muhith, U.; Irizarry-Cole, J.; Stolz, K.; Sanz, L.A.; Hartono, S.; et al. SETX (senataxin), the helicase mutated in AOA2 and ALS4, functions in autophagy regulation. Autophagy 2021, 17, 1889–1906. [Google Scholar] [CrossRef]
- Tanner, N.K.; Linder, P. DExD/H box RNA helicases: From generic motors to specific dissociation functions. Mol. Cell 2001, 8, 251–262. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Alzu, A.; Bermejo, R.; Begnis, M.; Lucca, C.; Piccini, D.; Carotenuto, W.; Saponaro, M.; Brambati, A.; Cocito, A.; Foiani, M.; et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012, 151, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Puget, N.; Lin, Y.L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533. [Google Scholar] [CrossRef]
- Grunseich, C.; Wang, I.X.; Watts, J.A.; Burdick, J.T.; Guber, R.D.; Zhu, Z.; Bruzel, A.; Lanman, T.; Chen, K.; Schindler, A.B.; et al. Senataxin Mutation Reveals How R-Loops Promote Transcription by Blocking DNA Methylation at Gene Promoters. Mol. Cell 2018, 69, 426–437.e427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentati, A.; Bejaoui, K.; Pericak-Vance, M.A.; Hentati, F.; Speer, M.C.; Hung, W.Y.; Figlewicz, D.A.; Haines, J.; Rimmler, J.; Ben Hamida, C.; et al. Linkage of recessive familial amyotrophic lateral sclerosis to chromosome 2q33-q35. Nat. Genet. 1994, 7, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Ben Hamida, M.; Hentati, F.; Ben Hamida, C. Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis). Conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain 1990, 113, 347–363. [Google Scholar] [CrossRef]
- Kress, J.A.; Kuhnlein, P.; Winter, P.; Ludolph, A.C.; Kassubek, J.; Muller, U.; Sperfeld, A.D. Novel mutation in the ALS2 gene in juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2005, 58, 800–803. [Google Scholar] [CrossRef]
- Siddiqi, S.; Foo, J.N.; Vu, A.; Azim, S.; Silver, D.L.; Mansoor, A.; Tay, S.K.; Abbasi, S.; Hashmi, A.H.; Janjua, J.; et al. A novel splice-site mutation in ALS2 establishes the diagnosis of juvenile amyotrophic lateral sclerosis in a family with early onset anarthria and generalized dystonias. PLoS ONE 2014, 9, e113258. [Google Scholar] [CrossRef]
- Sprute, R.; Jergas, H.; Olmez, A.; Alawbathani, S.; Karasoy, H.; Dafsari, H.S.; Becker, K.; Daimaguler, H.S.; Nurnberg, P.; Muntoni, F.; et al. Genotype-phenotype correlation in seven motor neuron disease families with novel ALS2 mutations. Am. J. Med. Genet. A 2021, 185, 344–354. [Google Scholar] [CrossRef]
- Eymard-Pierre, E.; Lesca, G.; Dollet, S.; Santorelli, F.M.; di Capua, M.; Bertini, E.; Boespflug-Tanguy, O. Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am. J. Hum. Genet. 2002, 71, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Sheerin, U.M.; Schneider, S.A.; Carr, L.; Deuschl, G.; Hopfner, F.; Stamelou, M.; Wood, N.W.; Bhatia, K.P. ALS2 mutations: Juvenile amyotrophic lateral sclerosis and generalized dystonia. Neurology 2014, 82, 1065–1067. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Vande Velde, C.; Eymard-Pierre, E.; Bertini, E.; Boespflug-Tanguy, O.; Cleveland, D.W. Unstable mutants in the peripheral endosomal membrane component ALS2 cause early-onset motor neuron disease. Proc. Natl. Acad. Sci. USA 2003, 100, 16041–16046. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef]
- Hadano, S.; Nichol, K.; Brinkman, R.R.; Nasir, J.; Martindale, D.; Koop, B.F.; Nicholson, D.W.; Scherer, S.W.; Ikeda, J.E.; Hayden, M.R. A yeast artificial chromosome-based physical map of the juvenile amyotrophic lateral sclerosis (ALS2) critical region on human chromosome 2q33-q34. Genomics 1999, 55, 106–112. [Google Scholar] [CrossRef]
- Otomo, A.; Hadano, S.; Okada, T.; Mizumura, H.; Kunita, R.; Nishijima, H.; Showguchi-Miyata, J.; Yanagisawa, Y.; Kohiki, E.; Suga, E.; et al. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 2003, 12, 1671–1687. [Google Scholar] [CrossRef] [Green Version]
- Topp, J.D.; Gray, N.W.; Gerard, R.D.; Horazdovsky, B.F. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 2004, 279, 24612–24623. [Google Scholar] [CrossRef] [Green Version]
- Nagano, I.; Murakami, T.; Shiote, M.; Manabe, Y.; Hadano, S.; Yanagisawa, Y.; Ikeda, J.E.; Abe, K. Single-nucleotide polymorphisms in uncoding regions of ALS2 gene of Japanese patients with autosomal-recessive amyotrophic lateral sclerosis. Neurol. Res. 2003, 25, 505–509. [Google Scholar] [CrossRef]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.I.; Ahmad, A.; Raza, S.I.; Amar, A.; Ali, A.; Bhatti, A.; John, P.; Mohyuddin, A.; Ahmad, W.; Hassan, M.J. In silico analysis of SIGMAR1 variant (rs4879809) segregating in a consanguineous Pakistani family showing amyotrophic lateral sclerosis without frontotemporal lobar dementia. Neurogenetics 2015, 16, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, Z.; Liu, L.; Xie, Y.; Zhan, Y.; Zi, X.; Wang, J.; Wu, L.; Xia, K.; Tang, B.; et al. A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 2015, 84, 2430–2437. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Epstein, M.L.; Andersen, K.A.; Ziskind-Conhaim, L.; Ruoho, A.E. The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience 2010, 167, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, J.; Shimohama, S.; Takano, S.; Harada, K.; Ueda, K.; Kimura, J. Novel G16S (GGC-AGC) mutation in the SOD-1 gene in a patient with apparently sporadic young-onset amyotrophic lateral sclerosis. Hum. Mutat. 1997, 9, 356–358. [Google Scholar] [CrossRef]
- Keckarevic, D.; Stevic, Z.; Keckarevic-Markovic, M.; Kecmanovic, M.; Romac, S. A novel P66S mutation in exon 3 of the SOD1 gene with early onset and rapid progression. Amyotroph. Lateral Scler. 2012, 13, 237–240. [Google Scholar] [CrossRef]
- Alexander, M.D.; Traynor, B.J.; Miller, N.; Corr, B.; Frost, E.; McQuaid, S.; Brett, F.M.; Green, A.; Hardiman, O. “True” sporadic ALS associated with a novel SOD-1 mutation. Ann. Neurol. 2002, 52, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Johnson, J.O.; Chia, R.; Miller, D.E.; Li, R.; Kumaran, R.; Abramzon, Y.; Alahmady, N.; Renton, A.E.; Topp, S.D.; Gibbs, J.R.; et al. Association of Variants in the SPTLC1 Gene With Juvenile Amyotrophic Lateral Sclerosis. JAMA Neurol. 2021. [Google Scholar] [CrossRef]
- Mohassel, P.; Donkervoort, S.; Lone, M.A.; Nalls, M.; Gable, K.; Gupta, S.D.; Foley, A.R.; Hu, Y.; Saute, J.A.M.; Moreira, A.L.; et al. Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nat. Med. 2021, 27, 1197–1204. [Google Scholar] [CrossRef]
- Daoud, H.; Zhou, S.; Noreau, A.; Sabbagh, M.; Belzil, V.; Dionne-Laporte, A.; Tranchant, C.; Dion, P.; Rouleau, G.A. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol. Aging 2012, 33, e835–e839. [Google Scholar] [CrossRef]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Pozner, T.; Regensburger, M.; Engelhorn, T.; Winkler, J.; Winner, B. Janus-faced spatacsin (SPG11): Involvement in neurodevelopment and multisystem neurodegeneration. Brain 2020, 143, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahed, A.C.; McDonough, B.; Gouvion, C.M.; Newell, K.L.; Dure, L.S.; Bebin, M.; Bick, A.G.; Seidman, J.G.; Harter, D.H.; Seidman, C.E. UBQLN2 mutation causing heterogeneous X-linked dominant neurodegeneration. Ann. Neurol. 2014, 75, 793–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunca, C.; Akcimen, F.; Coskun, C.; Gundogdu-Eken, A.; Kocoglu, C.; Cevik, B.; Bekircan-Kurt, C.E.; Tan, E.; Basak, A.N. ERLIN1 mutations cause teenage-onset slowly progressive ALS in a large Turkish pedigree. Eur. J. Hum. Genet. 2018, 26, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Adly, N.; Al Dhalaan, H.; Alkuraya, F.S. A nullimorphic ERLIN2 mutation defines a complicated hereditary spastic paraplegia locus (SPG18). Neurogenetics 2011, 12, 333–336. [Google Scholar] [CrossRef] [Green Version]
- Koroglu, C.; Yilmaz, R.; Sorgun, M.H.; Solakoglu, S.; Sener, O. GNE missense mutation in recessive familial amyotrophic lateral sclerosis. Neurogenetics 2017, 18, 237–243. [Google Scholar] [CrossRef]
- Carrillo, N.; Malicdan, M.C.; Huizing, M. GNE Myopathy: Etiology, Diagnosis, and Therapeutic Challenges. Neurotherapeutics 2018, 15, 900–914. [Google Scholar] [CrossRef] [Green Version]
- Harms, M.M.; Miller, T.M.; Baloh, R.H. TARDBP-Related Amyotrophic Lateral Sclerosis. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; GeneReviews® [Internet]: Seattle, WA, USA, 1993. [Google Scholar]
- Yamaura, G.; Higashiyama, Y.; Kusama, K.; Kunii, M.; Tanaka, K.; Koyano, S.; Nakashima, M.; Tsurusaki, Y.; Miyake, N.; Saitsu, H.; et al. Novel VRK1 Mutations in a Patient with Childhood-onset Motor Neuron Disease. Intern. Med. 2019, 58, 2715–2719. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.P.; Soeiro, E.S.M.; Silveira, F.; Pinto, S.; Gromicho, M.; Sousa, A.B.; Leao, M.; De Carvalho, M. VRK1 variants in two Portuguese unrelated patients with childhood-onset motor neuron disease. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2020, 21, 291–295. [Google Scholar] [CrossRef]
- Renbaum, P.; Kellerman, E.; Jaron, R.; Geiger, D.; Segel, R.; Lee, M.; King, M.C.; Levy-Lahad, E. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am. J. Hum. Genet. 2009, 85, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Toda, T.; Tsuji, S. Juvenile amyotrophic lateral sclerosis with complex phenotypes associated with novel SYNE1 mutations. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2020, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Nadaf, S.N.; Chakor, R.T.; Kothari, K.V.; Mannan, A.U. Synaptic Nuclear Envelope Protein 1 (SYNE 1) Ataxia with Amyotrophic Lateral Sclerosis-like Presentation: A Novel Synaptic Nuclear Envelope Protein 1 (SYNE 1) Gene Deletion Mutation from India. Ann. Indian Acad. Neurol. 2020, 23, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Hao, Y.; Cao, Z.; Zhou, C.; Zhang, J.; Wang, R.; Sun, S.; Gu, W. Autosomal Recessive Cerebellar Ataxia Type 1: Phenotypic and Genetic Correlation in a Cohort of Chinese Patients with SYNE1 Variants. Cerebellum 2021, 20, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Fan, D. A novel mutation of BICD2 gene associated with juvenile amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2017, 18, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Waldrop, M.A.; Wilson, R.K.; Flanigan, K.M. The Genotypic and Phenotypic Spectrum of BICD2 Variants in Spinal Muscular Atrophy. Ann. Neurol. 2020, 87, 487–496. [Google Scholar] [CrossRef]
- Wu, C.; Fan, D. A Novel Missense Mutation of the DDHD1 Gene Associated with Juvenile Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2016, 8, 291. [Google Scholar] [CrossRef] [Green Version]
- Schuurs-Hoeijmakers, J.H.; Geraghty, M.T.; Kamsteeg, E.J.; Ben-Salem, S.; de Bot, S.T.; Nijhof, B.; van de, V., II; van der Graaf, M.; Nobau, A.C.; Otte-Holler, I.; et al. Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2012, 91, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Liao, Y.H.; Ionita, C.; Bale, A.E.; Darras, B.; Acsadi, G. Mitochondrial Membrane Protein-Associated Neurodegeneration Mimicking Juvenile Amyotrophic Lateral Sclerosis. Pediatr. Neurol. 2016, 64, 83–86. [Google Scholar] [CrossRef]

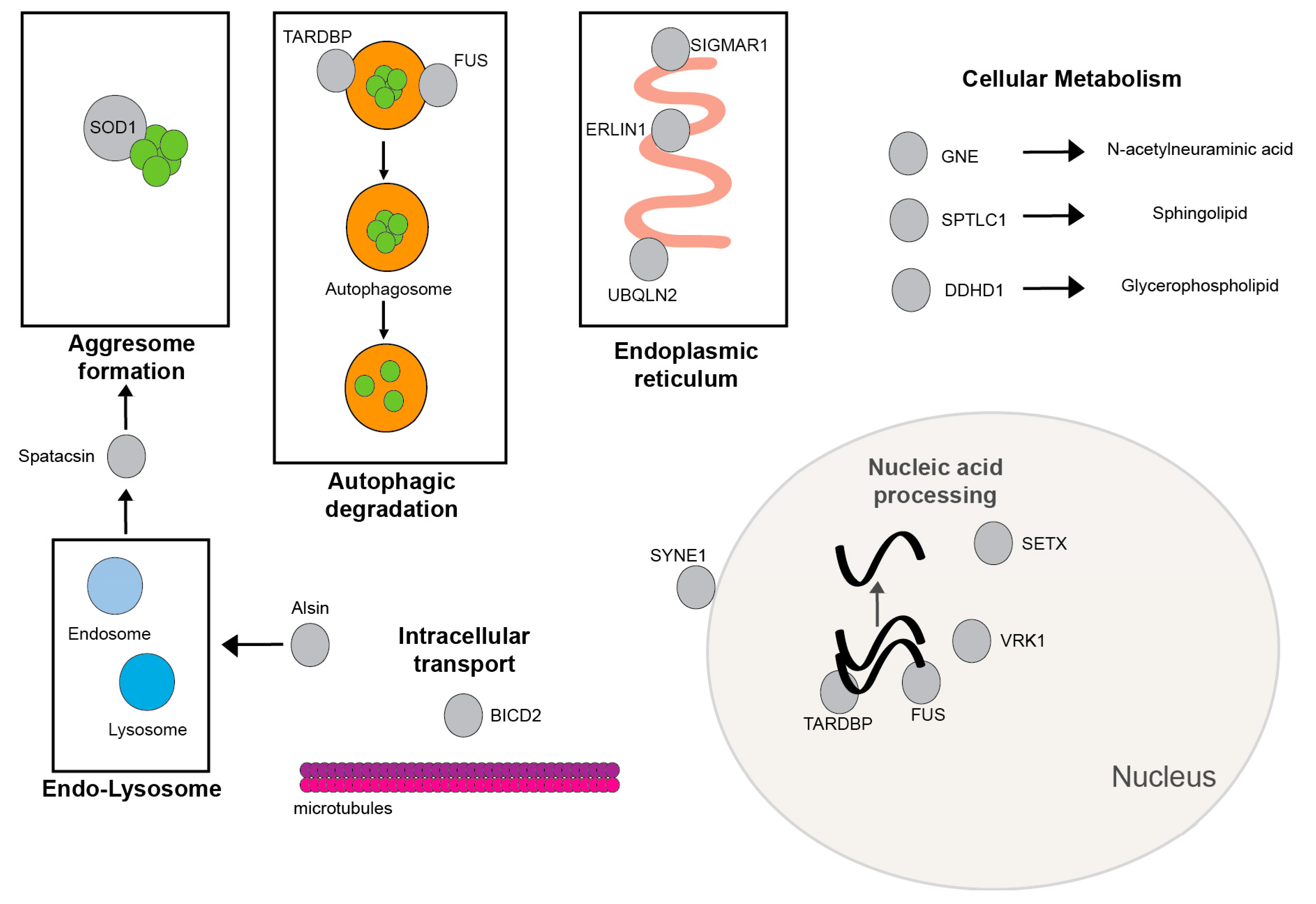

{kind=link}
{kind=link}
| GENE Mutation | FUS | SETX | ALS2 | SIGMAR1 | SOD1 | SPTLC1 | SPATACSIN | ||
| Inheritance pattern | De nova | AD | AR | AR | De nova | De nova and AD | AR | ||
| Coding effect | Het. | Het. | Hom. or complex Het. | Hom. | Het. | Het. | Hom. or complex Het. | ||
| Age of onset | Early 3rd decade | 2nd decade | Early 1st decade | Early 1st decade | Late 2nd decade | 1st–2nd decade | 2nd–3rd decade | ||
| Initial presentation | Bulbar onset | Distal leg weakness | Spasticity, dysarthria, dysphagia & bulbar weak | Spasticity & weakness in forearms | Asymmetric, early LMN signs | Spasticity, then LMN & bulbar involvement | Spasticity, distal weakness & bulbar involvement | ||
| Clinical progression | Rapid | Slow | Slow | Slow | Rapid | Slow | Slow | ||
| Other features | Myoclonic jerks, tremor | Cerebellar findings | Early anarthria, scoliosis | No bulbar or respiratory symptoms | None noted | Weight loss | None | ||
| Cognitive loss | Occasional | None | Pseudobulbar affect, no cognitive decline | None | None | None | None | ||
| EDX | Motor neuronopathy | Motor neuronopathy | Motor neuronopathy | Motor neuronopathy | Motor neuronopathy | Motor neuronopathy, sensory changes in one patient | Motor neuronopathy | ||
| MRI | Frontal cortical atrophy, pyramidal tract signal | NA | Mild cortical & spinal atrophy | Normal | Normal | NA | Normal | ||
| Other non-ALS associated disorders | None | Ocular Apraxia Type 2 | JPLS, HSP, dystonia | dHMN | None | HSAN | HSP | ||
| Selected references | Refs. [3,8,9] | Refs. [23,24,25] | Refs. [37,38,39] | Refs. [48,49,50] | Refs. [53,54,55] | Refs. [57,58] | Refs. [59,60] | ||
| GENE Mutation | UBQNL2 | ERLIN1 | GNE | TARDBP | VRI1 | SYNE1 | BICD2 | DDHD1 | |
| Inheritance pattern | X-linked dominant | AR | AR | - | AR | AR | De nova | AR | |
| Coding effect | X-linked dominant | Hom. | Hom. | Het. | Hom. or complex Het. | Hom. or complex Het. | Het. | Hom. | |
| Age of onset | 2nd decade | 2nd decade | 2nd–3rd decade | Early 3rd decade | 1st decade | 2nd decade | 2nd decade | 2nd decade | |
| Initial presentation | Details NA | Spasticity | Distal lower limb paraspinal, bulbar weakness | Distal upper limb weakness | Lower limb weakness & spasticity, later hand involvement. | Distal predominant weakness | Predominant UMN signs with tongue fasciculations | Details NA | |
| Clinical progression | Slow | Slow | Moderate | Rapid | Slow | Moderate | NA | NA | |
| Other features | None | None | None | None | Sensory | Dysarthria, dysphagia, mild limb ataxia | Dysarthria, dysphagia | None | |
| Cognitive loss | Dementia/FTD | NA | NA | None | Mild | Cognitive decline | Cognitive decline | Pseudobulbar affect | NA |
| EDX | Motor neuronopathy | Motor neuronopathy | Motor neuronopathy | Motor neuronopathy | Motor neuronopathy | Motor neuronopathy & sensory neuropathy | Motor neuronopathy | Motor neuronopathy & sensory neuropathy | |
| MRI | NA | NA | NA | NA | Normal | Mild cerebellar atrophy | Normal | Normal | |
| Other non-ALS associated disorders | FTD | HSP | Myopathy | None | Infantile SMA | Pure cerebellar ataxia | Distal SMA | HSP | |
| Selected references | Ref. [62] | Ref. [64] | Ref. [66] | Ref. [1] | Refs. [69,70] | Refs. [72,73] | Ref. [75] | Ref. [76] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehky, T.; Grunseich, C. Juvenile Amyotrophic Lateral Sclerosis: A Review. Genes 2021, 12, 1935. https://doi.org/10.3390/genes12121935
Lehky T, Grunseich C. Juvenile Amyotrophic Lateral Sclerosis: A Review. Genes. 2021; 12(12):1935. https://doi.org/10.3390/genes12121935
Chicago/Turabian StyleLehky, Tanya, and Christopher Grunseich. 2021. "Juvenile Amyotrophic Lateral Sclerosis: A Review" Genes 12, no. 12: 1935. https://doi.org/10.3390/genes12121935
APA StyleLehky, T., & Grunseich, C. (2021). Juvenile Amyotrophic Lateral Sclerosis: A Review. Genes, 12(12), 1935. https://doi.org/10.3390/genes12121935